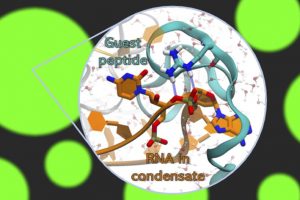
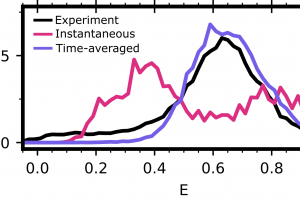
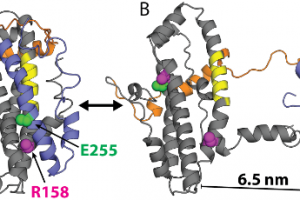
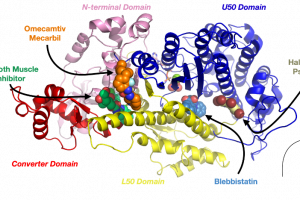
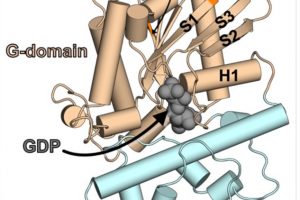
Improving protein design on Folding@home
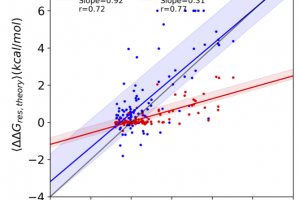
The protein design field has been making great strides with the advent of new machine learning tools trained on large databases of protein structures and sequences. While designs coming from…
Read more