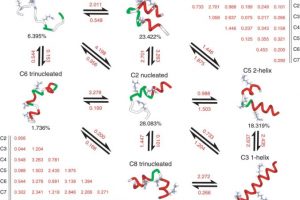
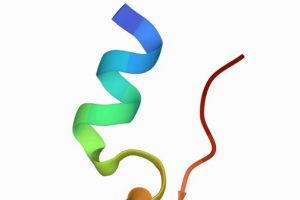
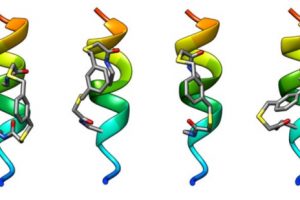
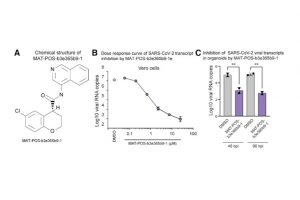
Understanding Druggable States in a Protein Implicated in Parkinson’s Disease
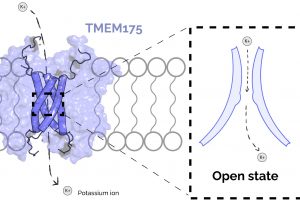
and Viktor Belay Section 1: How does the breakdown of a brain cell’s recycling plant lead to neurodegenerative disease? Cellular functions are dependent on the activity of a vast ensemble…
Read more