Help us reach our weekend goal so the COVID Moonshot can make rapid progress toward a COVID-19 therapy!
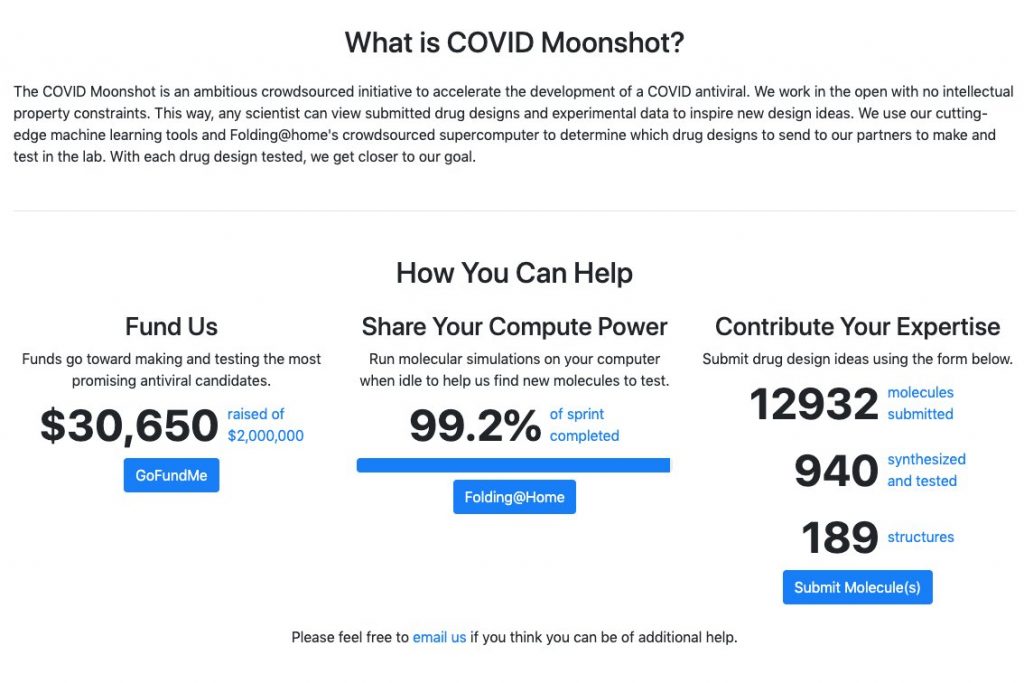
The First COVID Moonshot Weekly Sprint has just come to a close! While this first sprint took us longer than a week, we’re now on track to field new weekly sprints, and are optimistic that the Folding@home community is up to the task of rallying to get the Second COVID Moonshot Sprint done in a week! We’re aiming to complete enough work to send new molecules off to our synthetic chemistry partners at Enamine this weekend, starting the next sprint Sun 23 Aug.
This week, we’re evaluating a library of 8428 potential compounds that could be made from a common intermediate in one step using building blocks that Enamine has in stock:
These compounds can all be rapidly synthesized in parallel by Enamine chemists, who can make 10-12 a week. By picking the compounds with a best chance of improving the potency against the SARS-CoV-2 main viral protease (Mpro), we can help the COVID Moonshot rapidly progress toward potent inhibitors that could make their way into clinical trials.
These compounds all build off a key X-ray structure—one of the 191 new structures that DiamondMX has collected for the COVID Moonshot so far (that you can view in your browser right here thanks to talented computational chemistry guru Rachael Skyner at Diamond Light Source). These structures give us a map to how to build onto our lead compounds to achieve potency as we progress toward a drug. Here’s an overall picture of the Mpro binding site with a large peptidomimetic inhibitor N3 bound:
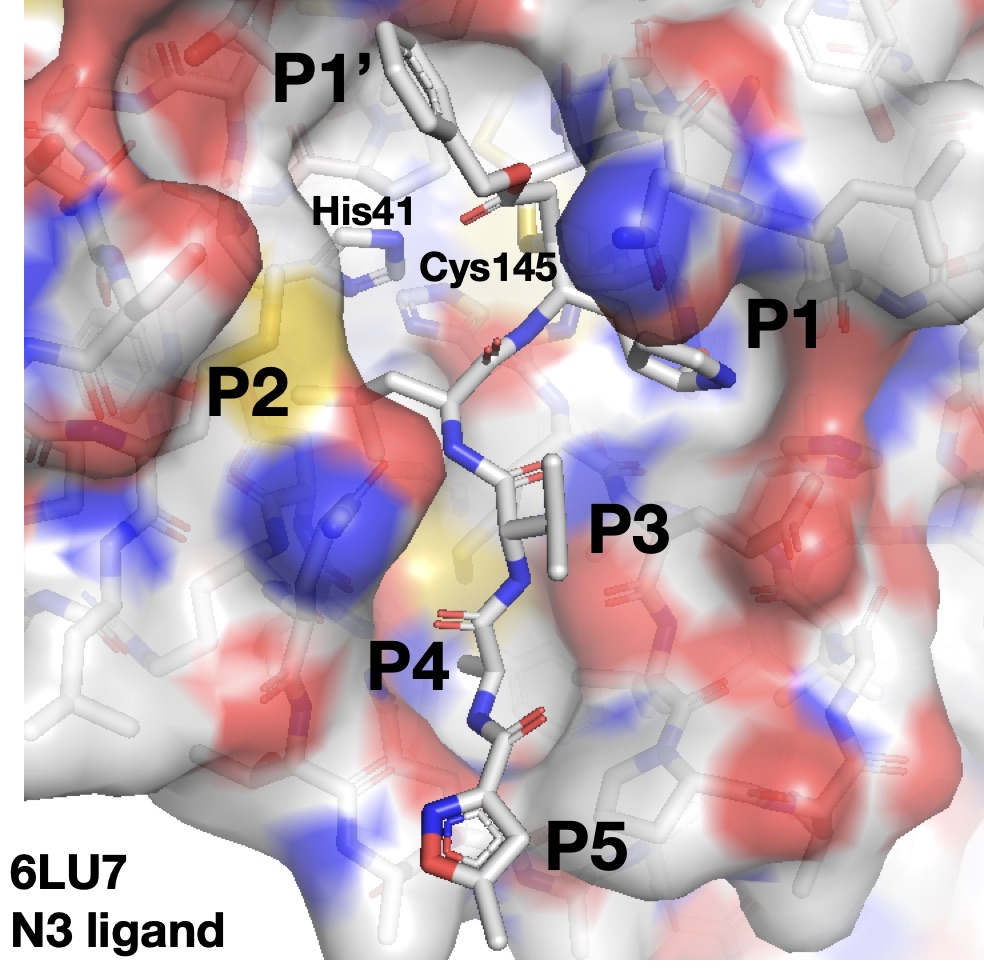
A good inhibitor will reach into as many pockets as possible to tightly bind to many of the crevices in Mpro and block off the active site, preventing the virus from maturing and providing a potent therapy or prophylactic for COVID-19. Unfortunately, peptidomimetics (very large small molecules that mimic peptides the enzyme normally binds) make very poor drugs, as it’s very difficult to optimize them to be bioavailable inside humans while not causing undesirable side effects. To give one example among many, the development of a peptidomimetic HIV inhibitor developed by Merck had to be restarted from scratch three times because of difficulties encountered during its development.
The quickest way to a drug is a small molecule that binds tightly enough to inhibit the protease with a much smaller number of atoms. One of the potent lead series uncovered among the thousands of crowdsourced COVID Moonshot submissions is 3-aminopyridine scaffold that binds tightly to pockets P1-P2:
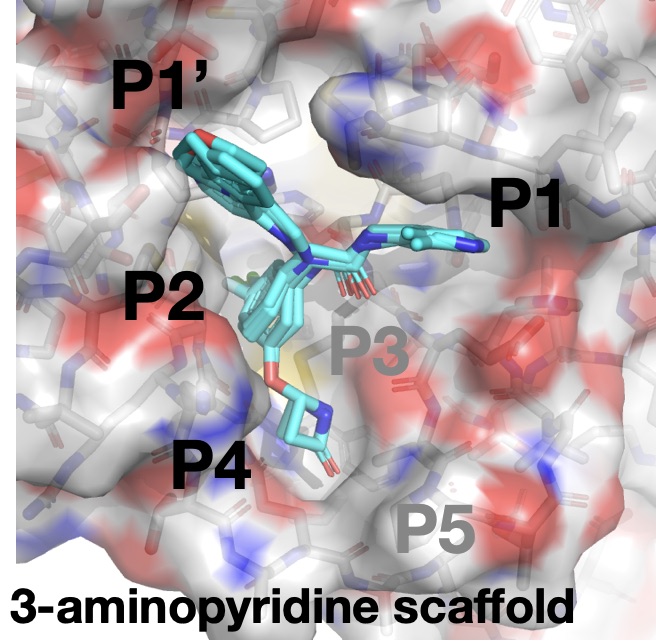
The scaffold itself shows a potency of 25 µM (micromolar), which means that at a concentration of just 25 µM of the inhibitor (lower is better!), 50% of the protease activity is inhibited! This is close enough to start optimizing. Surprisingly, the scaffold itself came from the clever merger of two molecules that nearly superimposed from the original DiamondMX/XChem fragment screen in March! Some of the other molecules that have been made since show it is possible to “grow” the molecule into the P1′ and P4 pockets, as well as tune the aminopyridine group reaching into the P1 pocket, to improve potency:
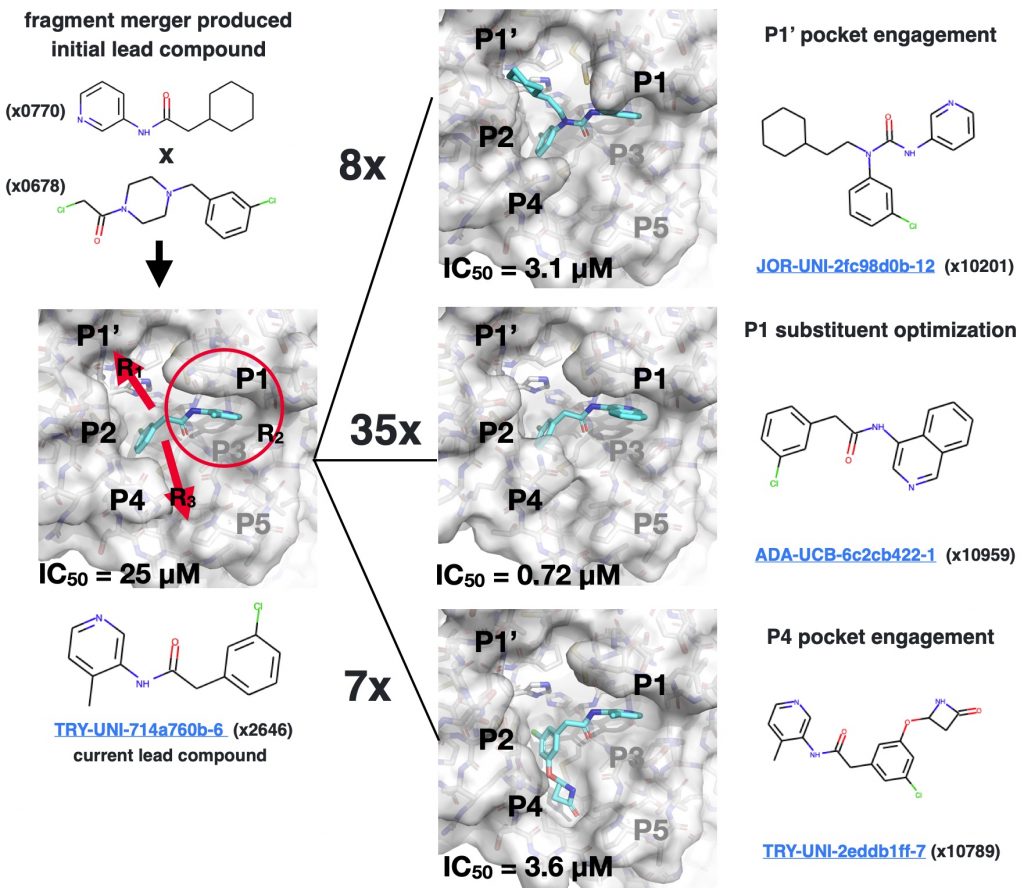
For this sprint, we’re trying to tune the chemical group pointing into the P4 pocket to see if we can pick up more than a 7x improvement in potency seen with the beta-lactam ring. We start with an X-ray structure with the beta-lactam nicely positioned in an opened P4 pocket:
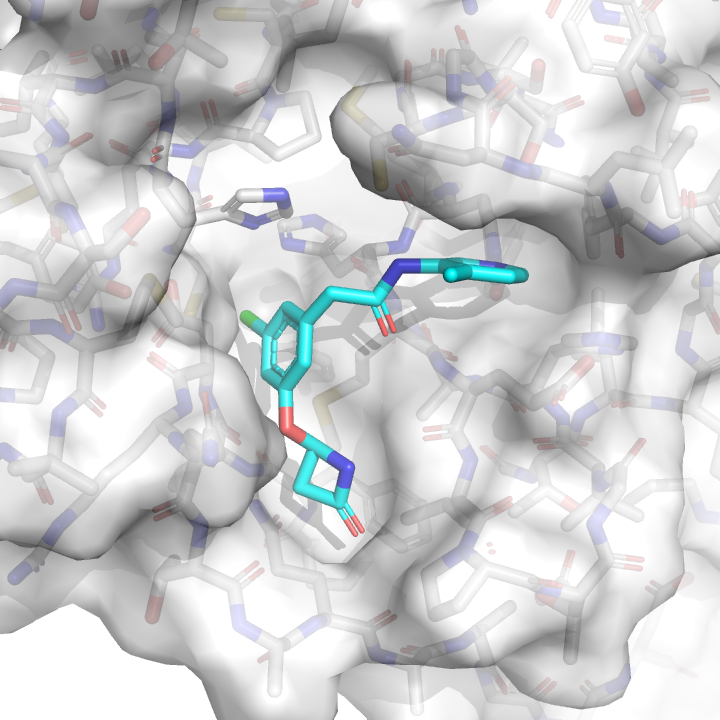
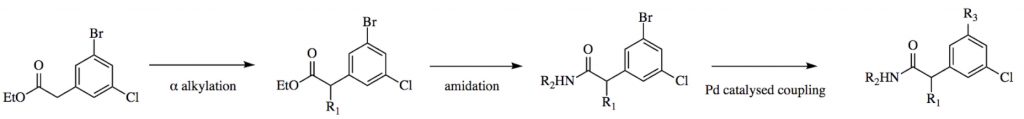
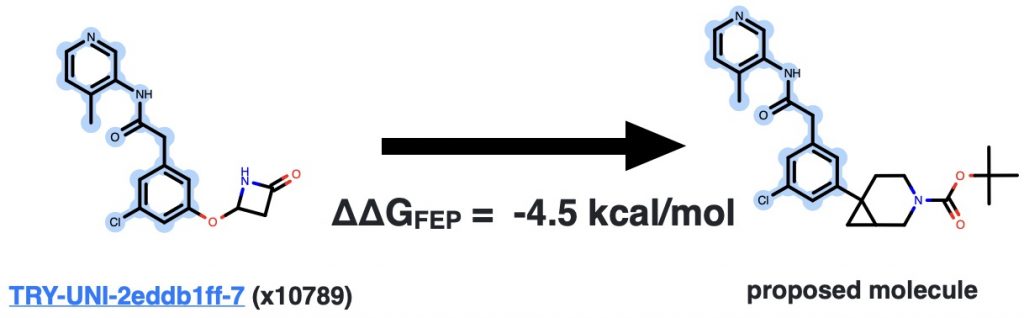
PostEra—a drug discovery technology company founded by Cambridge group leader Alpha Lee and founding member of the COVID Moonshot—used its machine learning platform, based on Molecule Transformer, to evaluate a multitude of synthetic routes to make modifications of our 3-aminopyridine lead compounts that could be rapidly synthesized by Enamine. They identified a short three-step synthetic route that can be used to install a variety of different chemical groups in the P4 pocket working from a common intermediate, which would allow Enamine chemists to make 10-12 molecules a week in pursuit of more potent compounds. However, we could make thousands of compounds given building blocks currently in stock. Which ones should we make?
That’s where Folding@home comes in! We build on an idea in this excellent blog post from the Practical Informatics blog from Pat Walters, who always provides code examples on GitHub! Keeping the scaffold atoms in common fixed to their locations in the X-ray structure, for each compound we could make, we identify a variety of different conformations using the OpenEye Omega toolkit*:
(*Full disclosure: JDC is on OpenEye’s Scientific Advisory Board)
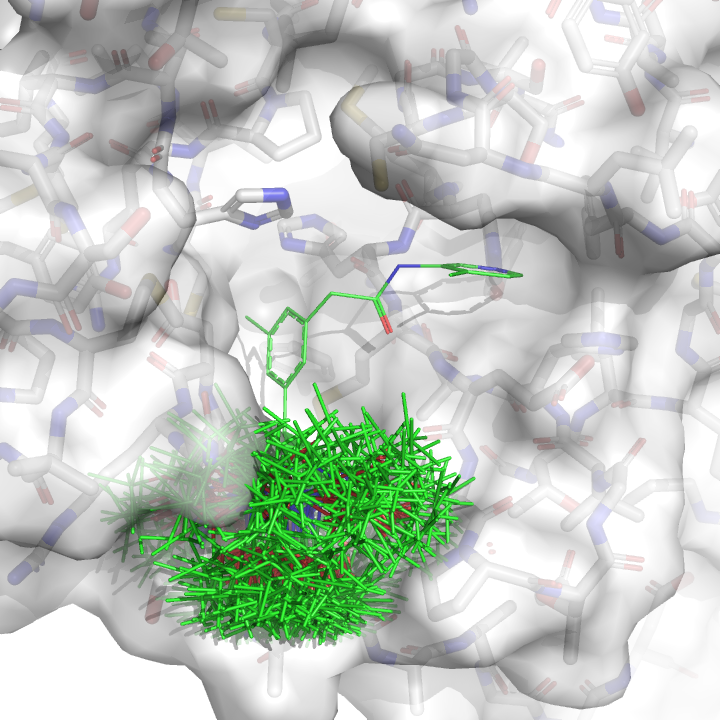
We then pick the conformation with the best docking score:
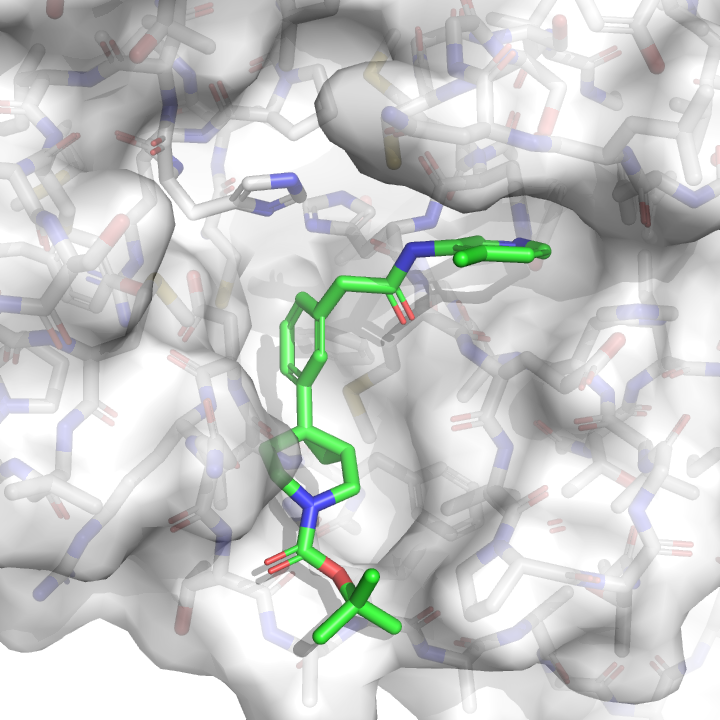
This conformation becomes the starting structure for relative alchemical free energy calculations, which we’ll explain in more detail in a future blog post. These calculations are expansive—we’re simulating each Mpro:inhibitor complex for a total of 1.2 microseconds of aggregate simulation time in this next sprint. That’s why we’re using the open source GPU-accelerated OpenMM core (core22) to squeeze as much performance out of Folding@Home volunteer machines as possible. At the end of the simulation, we produce an estimate of the free energy difference, where a negative number means the new compound is predicted to bind better:
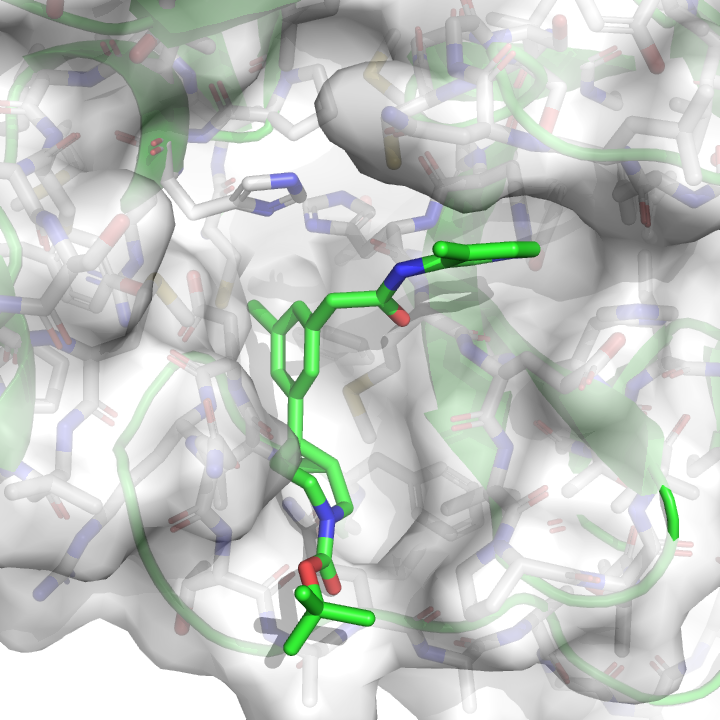
We also get snapshots of how we think the new molecule will bind, which can gratifyingly sometimes (but not always!) be very similar to how we expect from docking!
How reliable do we expect the calculations to be? We won’t know for sure until the first batch of compounds (which was sent to Enamine to start synthesizing this weekend) through our assays in 2-3 weeks and see how well the compounds bind and how they fit into the active site, but we can get an idea of what we expect using the retrospective performance on related compounds where we have already measured IC50s:
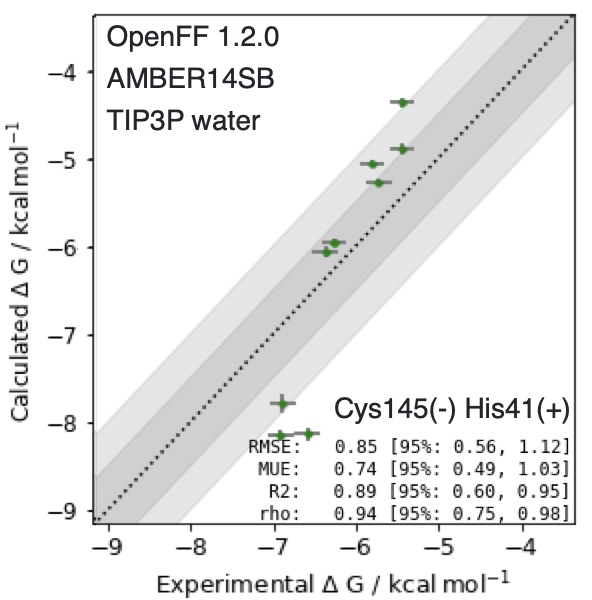
These calculations use the new Open Force Field Initiative OpenFF 1.2.0 force field (see slides), the product of a radical new open science, open source collaboration to develop high-quality free and open force fields for anyone to use for drug discovery. While there are only a few compounds in the retrospective set, the performance is astonishingly good: an RMSE of 0.85 [95% CI: 0.56, 1.12] kcal/mol, and a correlation coefficient rho of 0.94 [95% CI: 0.75, 0.98], suggesting these calculations work exceedingly well on this system. It’s important to keep in mind that we’re performing relative free energy transformations that involve disappearing and appearing many more atoms than are typical for these kinds of calculations (necessitating >1 microsecond/transformation), but we’re excited to see the results in the next 2-3 weeks, all of which will be posted to http://postera.ai/covid immediately after they become available.
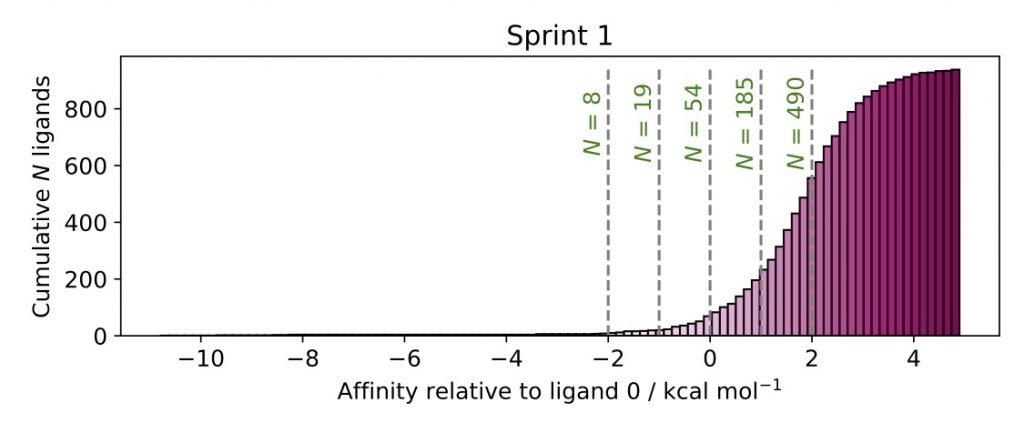
At the end of the first sprint, computational chemist Hannah Bruce Macdonald—whose awesome work on our open source software perses, together with Tri-Institutional PhD Program in Chemical Biology student Dominic Rufa made all of this possible—sorted the predicted compounds from best to worst, and surprisingly found only ~54 were predicted to bind better than the current lead compound:
Here are a few of the best compounds that emerged from the first sprint—shown in their docked poses, rather than their simulated poses:
Once these compounds have been synthesized and assayed, we’ll review how well we did!
In the meantime, we’ll be posting more about the COVID Moonshot’s progress, how we’re working to rapidly develop a new drug at lightning speed during a pandemic, and new data later this week! In the meantime, fire up those GPUs and help us reach 100% by the weekend! With your help, we can quickly discover potent molecules that shut down SARS-CoV-2 and find a safe, effective, and inexpensive way to treat and defeat COVID-19.
Help us help you
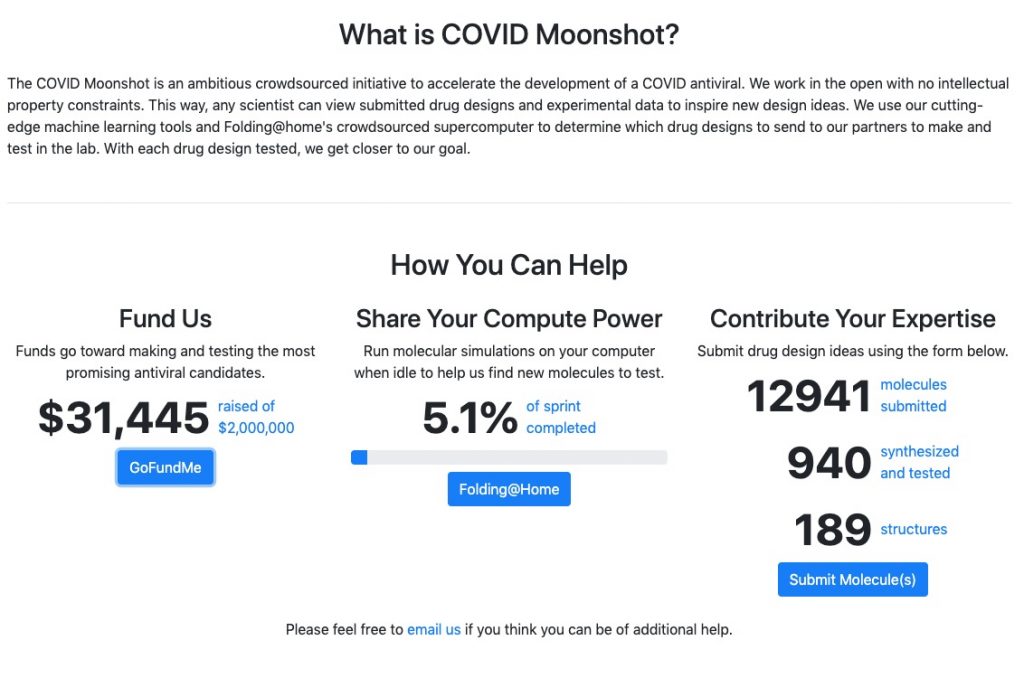
The COVID Moonshot is an open science, patent free collaboration to develop an inexpensive therapeutic for COVID-19. You can help us meet our goal by supporting our fundraiser to pay for the synthesis of new compounds: https://www.gofundme.com/f/covidmoonshot