This is an update on Folding@home’s efforts to assist researchers around the world taking up the global fight against COVID-19.
After initial quality control and limited testing phases, Folding@home team has released an initial wave of projects simulating potentially druggable protein targets from SARS-CoV-2 (the virus that causes COVID-19) and the related SARS-CoV virus (for which more structural data is available) into full production on Folding@home. Many thanks to the large number of Folding@home donors who have assisted us thus far by running in beta or advanced modes.
This initial wave of projects focuses on better understanding how these coronaviruses interact with the human ACE2 receptor required for viral entry into human host cells, and how researchers might be able to interfere with them through the design of new therapeutic antibodies or small molecules that might disrupt their interaction.
In the coming days, we hope to take advantage of some of the new structural biology and biochemical data that is being rapidly released by researchers around the world who are working to understand these viruses and strategies for defeating them. This work has been largely disseminated by preprint servers such as bioRxiv and chemRxiv, which aim to make research rapidly available to both other researchers and the public for other scientists to broadly evaluate and immediately start building on. We have also forged several new collaborations with other laboratories where we hope Folding@home will provide valuable support in COVID-19 research efforts.
While we will rapidly release the simulation datasets for others to use or analyze, we aim to look for alternative conformations and hidden pockets within the most promising drug targets, which can only be seen in simulation and not in static X-ray structures. We hope that these structures—once validated by emerging compound screening data—could help direct the virtual screening campaigns or the targeting of new pockets for which atomistic structures were not yet available.
Below, we provide short descriptions of the projects. Note that all input files are being made available on GitHub here for other researchers to take advantage of:
https://github.com/foldingathome/coronavirus
This repository will evolve over the coming days as we add more projects and documentation. We will start posting datasets with structures on publicly available servers as soon as we have useful data to report.
All projects are using the new GPU-accelerated Core22 based on the open source OpenMM biomolecular simulation engine.
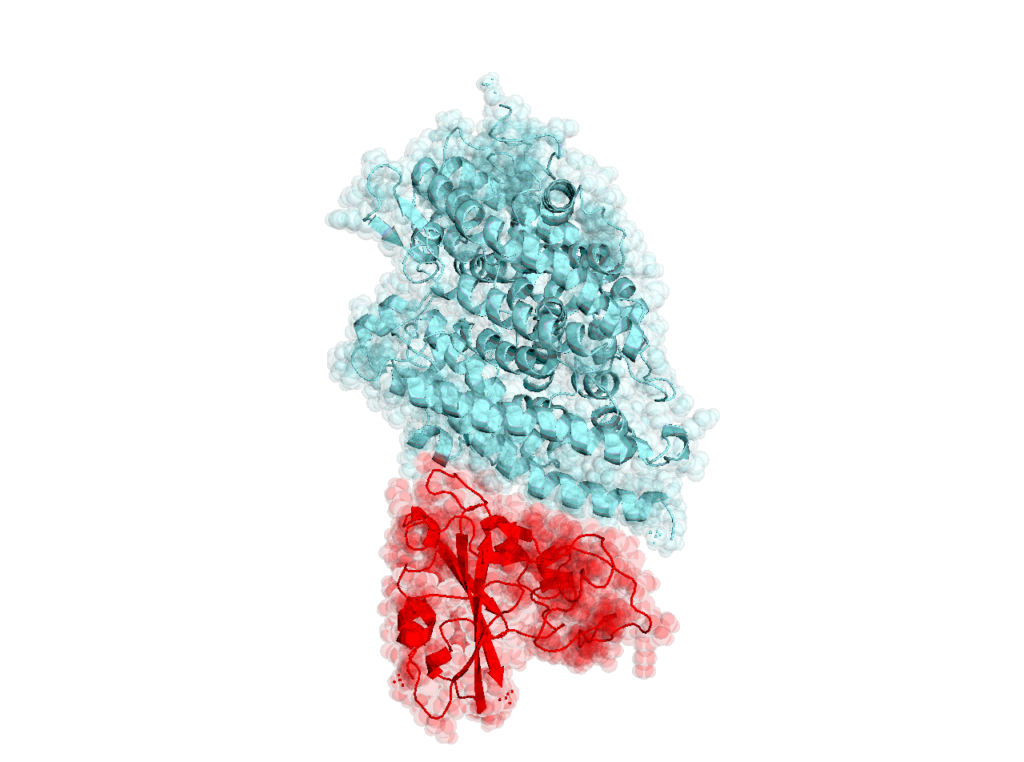
SARS-CoV-2 RBD domain in complex with human ACE2 receptor (PDBID: 6vsb, 6acg) [10.1126/science.abb2507, 10.1371/journal.ppat.1007236]
11741: Coronavirus SARS-CoV-2 (COVID-19 causing virus) receptor binding domain in complex with human receptor ACE2. atoms: 165550, credit: 15396
11746: Coronavirus SARS-CoV-2 (COVID-19 causing virus) receptor binding domain in complex with human receptor ACE2 (alternative structure to 11741). atoms: 182699, credit: 16615

SARS-CoV-2 main protease in complex with an inhibitor N3 (PDBID: 6lu7) [Not yet published]
11742: Coronavirus SARS-CoV-2 (COVID-19 causing virus) protease in complex with an inhibitor. atoms: 62227, credit: 9405
11743: Coronavirus SARS-CoV-2 (COVID-19 causing virus) protease – potential drug target. atoms: 62180, credit: 9405
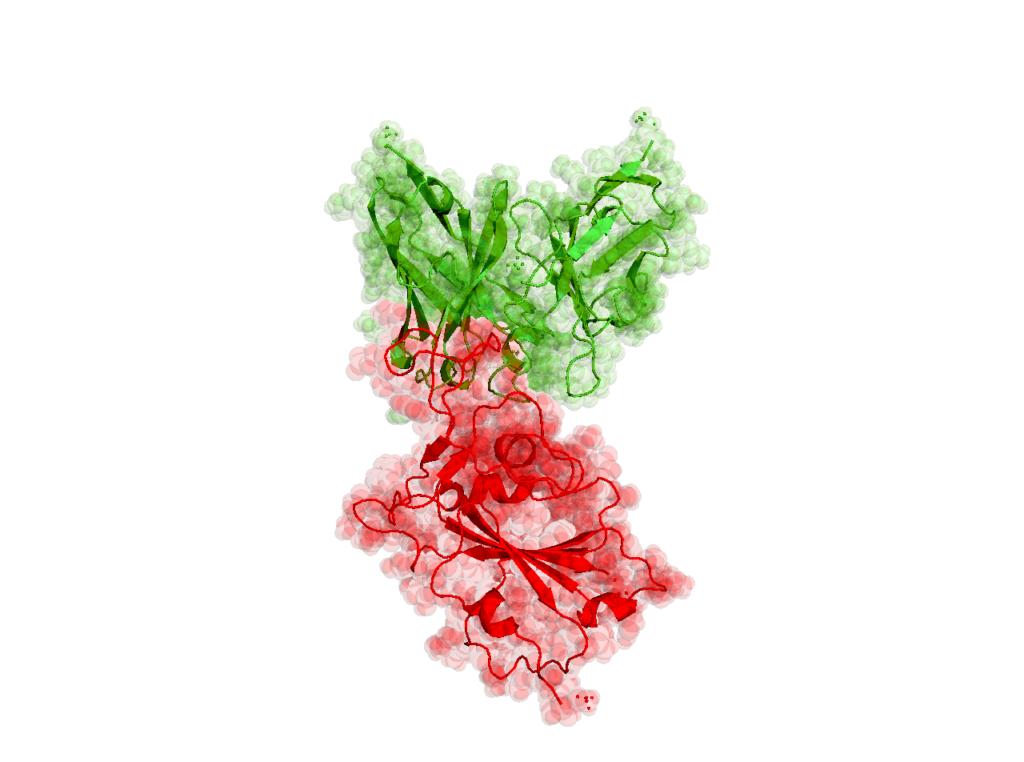
SARS-CoV-2 RBD domain in complex with human neutralizing S230 antibody Fab fragment (PDBIDs: 6nb7, 6nb8, 2ghv) [ 10.1016/j.cell.2018.12.028 (for both 6nb7 and 6nb8), 10.1074/jbc.M603275200]
11744: Coronavirus SARS-CoV (SARS causing virus) receptor binding domain trapped by a SARS-CoV S230 antibody. atoms: 109578, credit: 7608
11745: Coronavirus SARS-CoV (SARS causing virus) receptor binding domain mutated to the SARS-CoV-2 (COVID-19 causing virus) trapped by a SARS-CoV S230 antibody. atoms: 110370, credit: 7685
To contact us to discuss collaborations or data, please email us at foldingathome@choderalab.org
Special thanks to TBCP graduate student Rafal Wiewiora and CBM graduate student Ivy Zhang for their work in modeling these structures from existing experimental data and preparing these projects, and to all the Folding@home donors who help make this work possible!
~ The Chodera lab SARS-CoV-2 team and Folding@home Consortium ~