One of the big challenges with simulations of protein dynamics is getting enough data. That’s where Folding@home and all of the compute power that you volunteer to dedicate to running simulations proves invaluable!
Another major factor is the mathematical description of the relevant physics used to run the simulations, called a force field because it defines the forces that every atom in the system exerts on every other atom. No amount of data can make up for having a bad force field. On the flip side, one needs a lot of data to assess if a force field is working well.
One thing we do periodically is run simulations of the same proteins with a variety of force fields to see which give the best all-around performance. In the past, we’ve found that most modern force fields give similar results for nicely folded proteins. However, the field has found that for extremely dynamics proteins, often called intrinsically disordered proteins, many force fields perform quite poorly. This observation has driven the development of new force fields that are intended to work well for both nicely folded and disordered proteins.
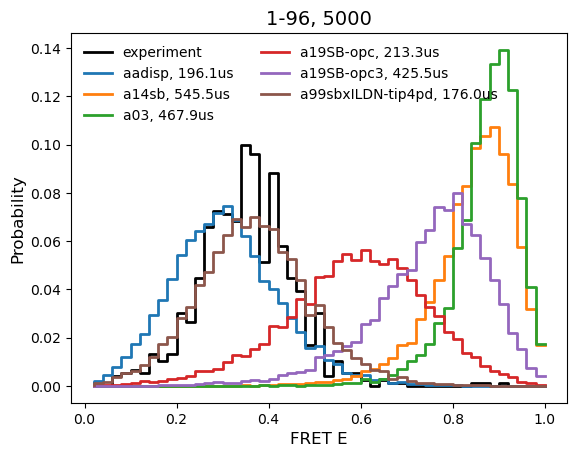
Over the past year, we’ve been performing extensive simulations of some disordered proteins with a variety of force fields and comparing to experiments called single molecule FRET measurement. Without going into great detail, these experiments report on the proximity of two dyes that are attached to different parts of the protein by measuring a phenomenon called energy transfer (E). We’ve recently developed powerful methods for predicting the distribution of E from large-scale simulations, providing a great opportunity to compare the distributions from simulations with those measured from experiments.
Encouragingly, some of the force fields developed to better model disordered proteins look really good! Our current favorite has quite a long name: amger99sb*-ildn with the tip4p (or tip4pd) water model. In the figure below you can see that predictions based on simulations using this force field agree quite well with experiment. In contrast, lots of force fields that work great for folded proteins do very poorly with this disordered protein, including amber03, amber14sb, and amber19sb. We’re also finding that the new force fields that perform well for disordered proteins continue to perform well for nicely folded proteins, so we plan to switch over to these force fields for all our future simulations.