A central goal of Folding@home is to use distributed computing to solve problems that would otherwise be intractable. Protein folding is a terrific problem to address with this approach: by spawning large numbers of short trajectories, we can sample rare events like folding that would otherwise require prohibitively long trajectories. More than twenty years ago (1), the first application of Folding@home was simply to sample rare folding events, a feat that was previously impossible (2). Later, the idea of rare-event sampling was expanded with a kinetic network framework called Markov models, to provide a complete description of the populations of different conformational states, and the rates at which states interconvert (3,4).
But modeling protein folding using only ensembles of short trajectories comes at a price! Since we are nearly always sampling trajectories out-of-equilibrium, we risk losing information about equilibrium populations. For example, a protein at room temperature may be in a folded state 99.9% of the time. That means if we wanted to very strictly generate trajectory data at equilibrium, using 1000 simulations at Folding@home, we would perform one simulation starting from an unfolded state, and 999 simulations from the folded state. That seems like a waste, if we are trying to learn about how a protein gets from an unfolded state to its folded state, right?
In a new paper from the Voelz lab, we address this problem using a recently-developed approach to building Markov models called multiensemble Markov models, or MEMMs (5). In this approach, biased simulations can be performed that encourage folding (or unfolding). The effects of this bias can then be removed, to build a Markov model of the folding reaction that correctly captures both the kinetics and the thermodynamics (equilibrium populations) of a protein.
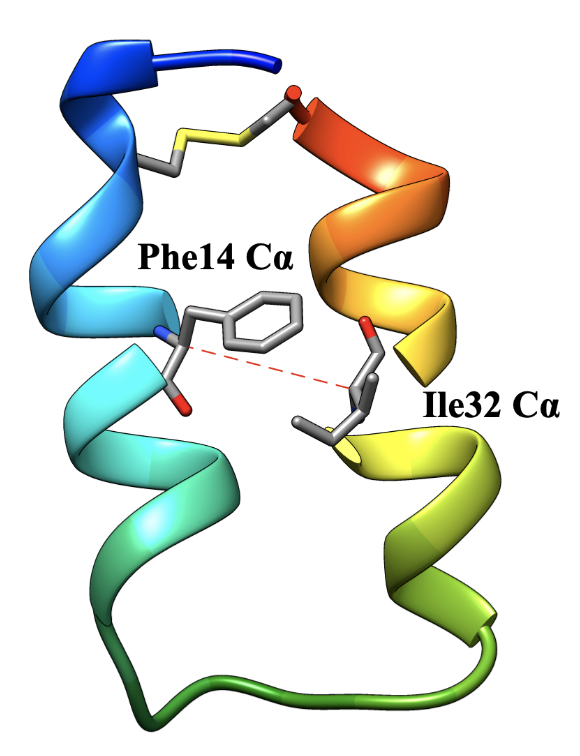
We use the MEMM approach to model the folding reaction of a small two-helix protein called Z34C. At room temperature, this protein has a folding time of about 10 µs, and an unfolding time of nearly 1 ms. When we performed unbiased simulations, few trajectories starting from unfolded states actually fold, and no trajectories starting from the folded state unfold. After adding biased simulations to our dataset (harmonic restraints on the distance between the alpha-carbons of Phe14 and Ile32), we find that we can sample folding and unfolding events, and obtain MEMMs that predict folding rates and populations in good agreement with experiments.
In the future, we believe approaches like this will be essential to modeling and designing protein stability and kinetics. You can read more about this work in our new manuscript:
Improved Estimates of Folding Stabilities and Kinetics with Multiensemble Markov Models. Si Zhang, Yunhui Ge, and Vincent A. Voelz. Biochemistry, Accepted (Oct 28, 2024).
https://doi.org/10.1021/acs.biochem.4c00573
References
1. Voelz, V. A., Pande, V. S., and Bowman, G. R. (2023) Folding@home: Achievements from over 20 years of citizen science herald the exascale era. Biophysical Journal S0006349523002011.
2. Snow, C. D., Nguyen, H., Pande, V. S., and Gruebele, M. (2002) Absolute comparison of simulated and experimental protein-folding dynamics. Nature 420, 102–106.
3. Pande, V. S., Beauchamp, K., and Bowman, G. R. (2010) Everything you wanted to know about Markov State Models but were afraid to ask. Methods 52, 99–105.
4. Voelz, V. A., Bowman, G. R., Beauchamp, K., and Pande, V. S. (2010) Molecular Simulation of ab Initio Protein Folding for a Millisecond Folder NTL9(1−39). J. Am. Chem. Soc. 132, 1526–1528.
5. Wu, H., Paul, F., Wehmeyer, C., and Noé, F. (2016) Multiensemble Markov models of molecular thermodynamics and kinetics. Proc Natl Acad Sci USA 113, E3221–E3230.