Our new work shows how to predict the results of single molecule experiments from large simulations, like those performed on Folding@home.
Showing that simulations are consistent with experiments is important for establishing the credibility of simulations and adding weight to any predictions that they lead to. Connecting with experiments performed on a single protein at a time (called a single molecule experiment) is a particularly compelling goal since these experiments are very similar in nature to watching a single protein in a simulation. Unfortunately, simulations and single molecule experiments have tended to give pretty different pictures of how a protein behaves, suggesting that predictions from simulations should be taken with a large grain of salt.
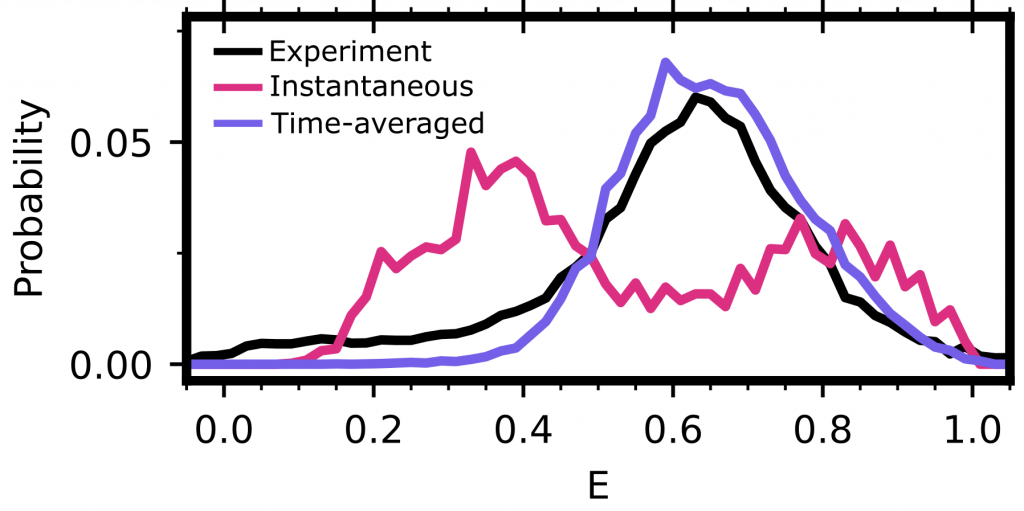
In this paper, we show that simulations and single molecule experiments are in much better agreement than one might have thought based on past work. The key insight is that single molecule experiments do not give you instantaneous feedback on how a protein is behaving. Instead, they report an average of what the protein looks like over a one to ten millisecond time interval. Accounting for the averaging across the dynamics that occur during this time (called time-averaging) makes a huge difference in how well predictions from simulations agree with experiments, as shown in the figure below. The key takeaway is that the curves from experiments and time-averaged predictions from our simulations are in good agreement, while the instantaneous prediction from our simulations is way off.