and Viktor Belay
Section 1: How does the breakdown of a brain cell’s recycling plant lead to neurodegenerative disease?
Cellular functions are dependent on the activity of a vast ensemble of macromolecular machines, including many of the proteins that we’ve discussed on this site as well as organelles like mitochondria. As cells get older, these machines break down, become waste, and need to be recycled or removed. This happens via a biochemical process called autophagy (literally, “self-eating”). Organelles called lysosomes play a central role in this process. These are small, highly acidic membrane-bound compartments within cells that mediate internal waste processing and removal. Lysosomes use a set of important proteins to dynamically sense the status of internal cellular waste accumulation and regulate waste removal (Figure 1) [1].
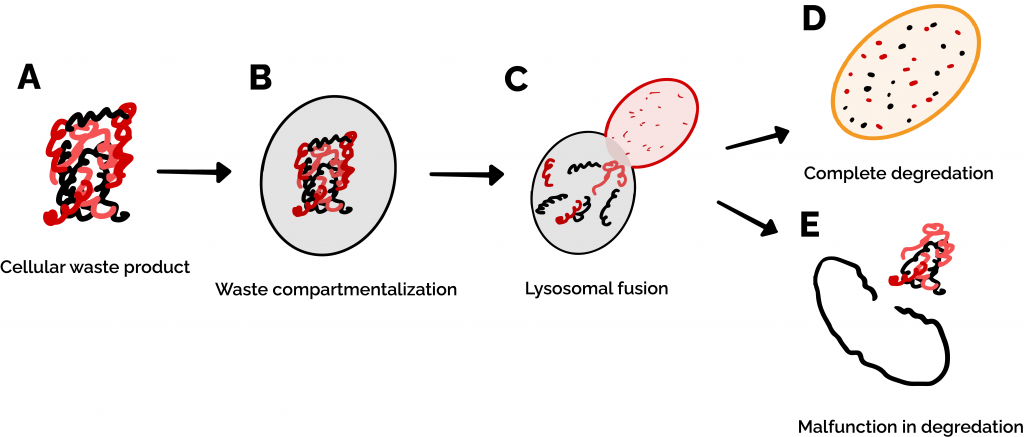
Figure 1: Autophagy is the process by which cells recycle proteins and organelles when they break down. (A) When these waste products build up in a cell, (B) they are compartmentalized by a lipid bilayer membrane called an autophagosome. (C-D) The autophagosome begins fusion with a lysosome, which chews up the waste products. (E) In the case that a process in the autophagy pathway malfunctions, cellular waste remains undegraded and continues to accumulate within and outside of the cell.
Many neurodegenerative diseases (such as Alzheimer’s and Parkinson’s) can be linked to a breakdown in autophagy, resulting in waste buildup and subsequent neuronal death [2]. Neurons in the brain are particularly vulnerable to a breakdown in autophagy. While other types of cells are regularly replaced by new ones and therefore never have a critical buildup of waste products, neurons cannot be easily replaced, and therefore have to rely on autophagy to clear out broken cellular machinery [1,2]. Additionally, neurons are constantly releasing and absorbing signals (such as when your brain tells your arm muscles to move!) which requires heavy production and recycling of a variety of cellular components. On top of this, you may have heard that the brain is the hardest-working ‘muscle’ in the body since it uses the most energy (in the form of food) of any organ. This energy requirement puts a lot of stress on mitochondria – the ‘powerhouse’ energy-producing organelles of the cell – causing them to break down rapidly, requiring your cells to recycle them and produce new ones at a rapid rate [3]. All of these factors mean that even a moderate decrease in the function of neuronal lysosomes can cause waste products to aggregate within these cells at a faster rate over the course of a person’s lifetime, leading to neurodegenerative diseases like Parkinson’s.
Section 2: What role does TMEM175 play in all this?
Transmembrane Protein 175 (TMEM175) is a protein in the lysosomal membrane that functions as a “channel” for potassium ions to move between the open space in the cell (the cytoplasm) and the space within the lysosome (the lysosomal lumen) (Figure 2 A) [5]. TMEM175 is thought to play an important role in autophagy as a lysosomal protein, but its exact function in this process is not yet clear. Nevertheless, dysfunction of TMEM175 is implicated in the development of Parkinson’s. We know this because several mutational variants of TMEM175 have been identified in Parkinson’s patients, and experiments have shown that mice with these variants have some of the molecular and behavioral hallmarks of Parkinson’s [4]. An understanding of the fundamental mechanisms of TMEM175 will lead to new ways to discover drugs or therapeutic interventions to treat, prevent, or potentially even reverse Parkinson’s Disease.
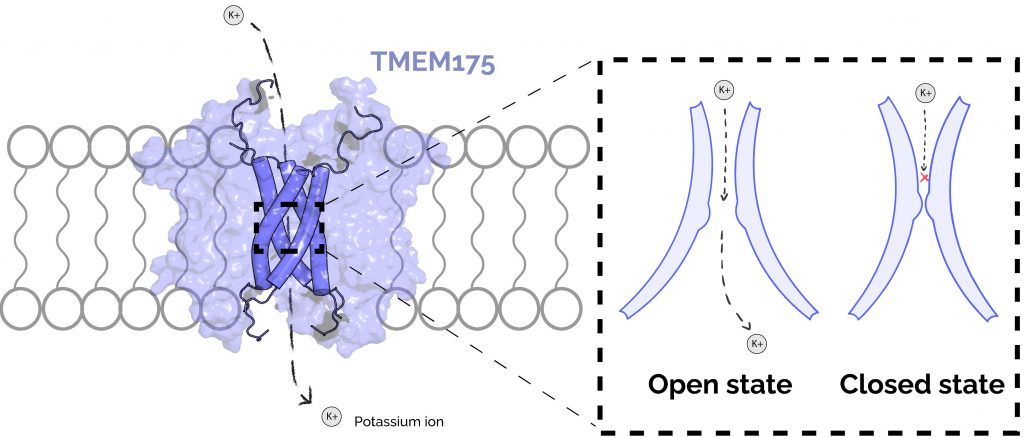
Figure 2: TMEM175 forms an ion channel in the lysosomal membrane and cycles through at least two structural states to translocate ions. An ion channel is essentially a pore in the membrane which allows only small charged molecules to pass through. (A) The TMEM175 pore is formed by 4 membrane-spanning helices (shown as dark-blue cylinders). The remainder of the TMEM175 protein (light-blue surface) maintains structure in the membrane and engages conformational change in response to stimuli. TMEM175 moves potassium (K+) ions between the cytoplasm of the cell and the lysosomal lumen. (B) TMEM175 regulates movement of potassium with a tight constriction within its pore. In the open state, this constriction dilates such that potassium ions are able to pass. In the closed state, the constriction is too small for potassium ions to pass through.
As an ion channel, one of the primary functions of TMEM175 is to facilitate the movement of ions into the lysosome. While it is not known how ion conduction by TMEM175 contributes to the development of Parkinson’s, variants have been identified in humans that alter the likelihood that one will develop the disease [6]. You may know that people can inherit variations in the DNA sequence that codes for a particular gene. In the case of TMEM175, this results in a change in the amino acid composition of the protein that has a functional effect. For example, some families have a variant of TMEM175 which predisposes them to develop Parkinson’s [6], while others have a “protective” variant that reduces the likelihood of developing Parkinson’s [4]. Notably, these variants also alter the ability of TMEM175 to conduct ions, suggesting that its ability to conduct ions contributes to its role in disease (Figure 3). Taken together, these results indicate that the ability for TMEM175 to allow potassium ions to move across the membrane is related to its role in maintaining healthy lysosomes and preventing Parkinson’s. This means that developing drugs which increase TMEM175 activity could mimic the effect of the protective variant in people and could result in a new therapy for patients with Parkinson’s Disease.
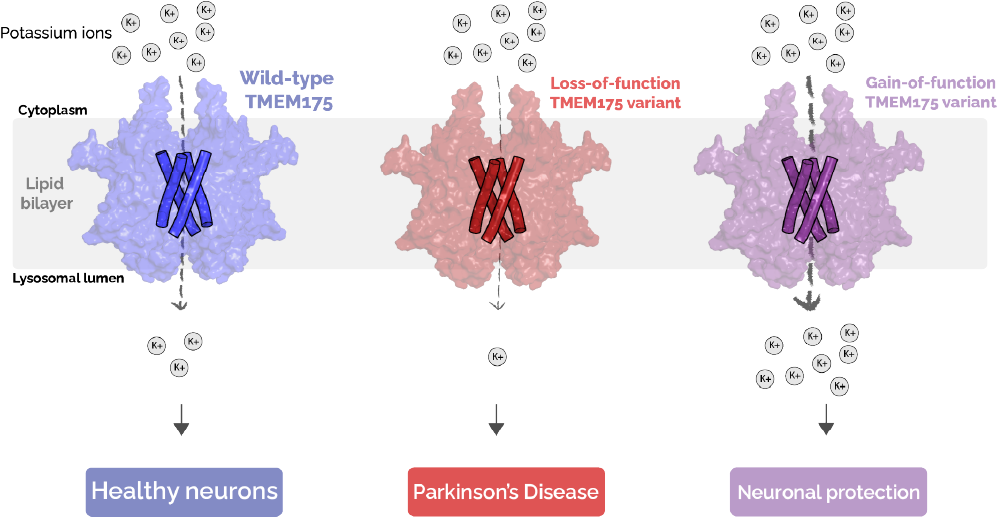
Figure 3: Inherited variants of TMEM175 have different activities and are associated with different outcomes. Loss-of-function variants of TMEM175 (middle) conduct less potassium ions relative to the typical, “wild-type” TMEM175 (left) and are associated with an increased likelihood of developing Parkinson’s. Gain-of-function variants of TMEM175 (right) conduct more potassium ions relative to “wild-type” TMEM175 and have been shown to reduce your likelihood of developing Parkinson’s.
To gain a better understanding of TMEM175 function, the molecular structure of human TMEM175 was resolved by a technique called cryogenic electron microscopy (“cryo-EM”). These experiments revealed two states of the protein: one that is shaped in a way that would allow for ion movement (the “open” state), and another is not (the “closed” state) [7]. These structures show that there is a tight constriction in the protein’s core that prevents ion movement in the closed state. In the open state, this constriction is dilated such that ions have enough room to pass through (Figure 2 B). However, because these structures are only static snapshots of the protein’s true configurational dynamics, they are missing the full picture of how the protein is activated and what states of the protein are amenable to novel drug design.
Section 3: How do we aim to better understand TMEM175?
Understanding the conformational landscape of TMEM175 will help us identify key targetable states, critically informing the design and validation of a new class of Parkinson’s Disease therapeutics. In order to better understand the molecular features that are important for TMEM175 opening, we will simulate both the open and closed states of the channel that were obtained from cryo-EM experiments. There is a problem though: TMEM175 is a relatively large protein, and the open-to-closed transition likely happens on long timescales. These attributes make our proposed simulations challenging to achieve on regular computers or even traditional supercomputers. This is where Folding@home (F@h) comes in! By using the distributed computing power of F@h, we will be able to simulate TMEM175 for timescales that are appropriate for observing the open-to-closed state transition in a reasonable amount of time.
Understanding the conformational landscape of TMEM175 will help us identify key targetable states, critically informing the design and validation of a new class of Parkinson’s Disease therapeutics.
Using F@h, we are simulating the motion of both the open and closed TMEM175 structures (Project 18500). Once we finish running the simulations, we will need to analyze the data – which is quite a challenge in itself! The simulations that we have discussed here will generate massive amounts of highly multi-dimensional data. Each atom in TMEM175 has a 3-dimensional position (x, y, z), and TMEM175 is composed of about 40,000 total atoms – that’s 3 x 40,000 = 120,000 total coordinates for a single simulation timestep. If we save a snapshot every nanosecond (10^-9 s) for an entire millisecond (10^-3 s) of simulation time, we will end up with ~10^11 total atomic coordinates at the end of the simulation. These raw data are clearly far beyond the scope of human interpretability. How then can we use these data to draw biologically and medically relevant conclusions?
An easy way to begin analysis of such a daunting set of simulation data is to transform the x,y,z positions of every atom into a smaller set of features that are easier to understand and plot. For simulation data, a popular method of dimensionality reduction is known as Time-Structure Independent Component Analysis (tICA) [8]. This method helps find the set of atomic motions that take the longest to complete over the course of the simulation. Another method we may use, called CARDS, was developed by Sukrit Singh in the Bowman lab which helps to identify regions of the protein whose motions are correlated to each other [9]. This could be useful in determining parts of the protein which play a key role in keeping the gate shut. Finally, machine-learning-based methods from the Bowman (DiffNets) and Delemotte (demystifying) labs could help us determine which features distinguish between the open and closed states of the protein [10, 11].
We can then use this knowledge to inform the design of a totally new molecule that could bind to the open state of the channel (and not the closed state), increasing its activity. This kind of molecule would be useful for studying TMEM175 function and could also lead to a new life-saving Parkinson’s Disease drug!
References
1. Glick, D., Barth, S. & Macleod, K. F. Autophagy: cellular and molecular mechanisms. The Journal of Pathology 221, 3–12 (2010).
2. Lie, P. P. Y. & Nixon, R. A. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiology of Disease 122, 94–105 (2019).
3. Vidyadhara, D. J., Lee, J. E. & Chandra, S. S. Role of the endolysosomal system in Parkinson’s disease. Journal of Neurochemistry 150, 487–506 (2019).
4. Wie, J. et al. A growth-factor-activated lysosomal K+ channel regulates Parkinson’s pathology. Nature 591, 431–437 (2021).
5. TMEM175 Is an Organelle K+ Channel Regulating Lysosomal Function – ScienceDirect. https://www-sciencedirect-com.ezproxy.med.cornell.edu/science/article/pii/S0092867415009782.
6. Jinn, S. et al. Functionalization of the TMEM175 p.M393T variant as a risk factor for Parkinson disease. Human Molecular Genetics 28, 3244–3254 (2019).
7. Oh, S., Paknejad, N. & Hite, R. K. Gating and selectivity mechanisms for the lysosomal K+ channel TMEM175. eLife 9, e53430 (2020).
8. Naritomi, Y. & Fuchigami, S. Slow dynamics in protein fluctuations revealed by time-structure based independent component analysis: The case of domain motions. J. Chem. Phys. 134, 065101 (2011).
9. Singh, S. & Bowman, G. R. Quantifying Allosteric Communication via Both Concerted Structural Changes and Conformational Disorder with CARDS. J. Chem. Theory Comput. 13, 1509–1517 (2017).
10. Ward, M. D. et al. Deep learning the structural determinants of protein biochemical properties by comparing structural ensembles with DiffNets. Nat Commun 12, 3023 (2021).
11. Fleetwood, O., Kasimova, M. A., Westerlund, A. M. & Delemotte, L. Molecular Insights from Conformational Ensembles via Machine Learning. Biophys J 118, 765–780 (2020).