TL;DR: During 2020, Folding@home was like the citizens’ fleet in the movie Star Wars: The Rise of Skywalker. Everyone came together to face a common foe, in this case the SARS-CoV-2 virus responsible for the COVID-19 pandemic. We are extremally grateful to everyone who joined in the fight, helping us to discover new antiviral candidates now in animal testing and publish four papers so far: 1) SARS-CoV-2 Simulations Go Exascale to Capture Spike Opening and Reveal Cryptic Pockets Across the Proteome, 2) SARS-CoV-2 Nsp16 activation mechanism and a cryptic pocket with pan-coronavirus antiviral potential, 3) The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA, 4) COVID Moonshot: Open Science Discovery of SARS-CoV-2 Main Protease Inhibitors by Combining Crowdsourcing, High-Throughput Experiments, Computational Simulations, and Machine Learning. We look forward to continuing to work with you, and wish you a happy new year!

It has certainly been an eventful year for everyone, and for Folding@home in particular.
The year started off with exciting developments in a number of directions. At the time, about 30K devices were participating in Folding@home. Together, we made great progress on understanding how small changes to proteins called myosin motors alter their function. Small changes to these motors are one of the most common causes of inherited diseases, so this first work is an important step towards being able to predict whether a newly discovered myosin variant is likely to cause disease or not. We also discovered new opportunities for combating Ebola virus, and helped repurpose an existing drug to combat the SFTS virus.
Then, like everyone else, our world got turned upside down by the COVID-19 pandemic.
In response, we quickly pivoted to focus all of our attention on the SARS-CoV-2 virus that is responsible for COVID-19 disease.
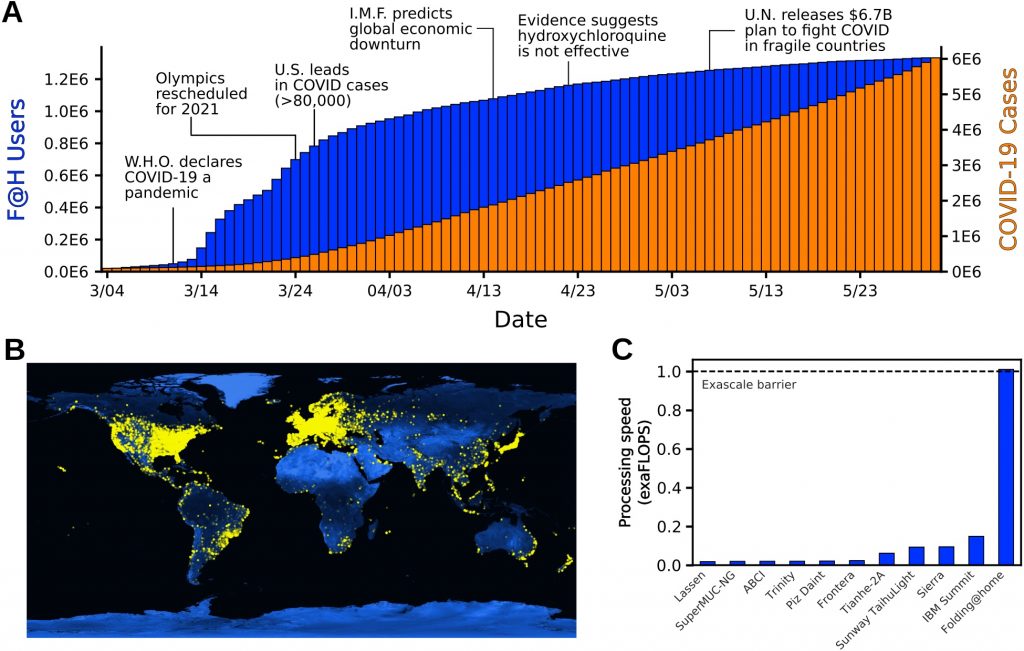
The public response to the announcement of our first simulations was massive, quickly turning Folding@home into the most powerful computer in the world. Within two weeks, over 400K new devices had joined the project, and we continued to grow rapidly from there. At our peak, over 280K GPUs and 4.8M CPU cores were helping to simulate as many proteins as possible from the SARS-CoV-2 virus, looking for new opportunities to combat the virus and new drugs to capitalize on these opportunities. By a very conservative estimate of the aggregate compute power of all these machines, Folding@home became the first exascale computer, having over 5-fold greater performance than the world’s fastest supercomputer at the time (the Summit supercomputer, which clocks in at 200 petaFLOPS). Using this compute power, we generated over 0.1 seconds of simulation data. That’s over 100,000 times more data than a typical simulation paper. Internally, we joked that we may have generated more simulation data than has been produced in the history of science, but the joke may actually be true!
One of our major foci was on the spike, which is one of the primary targets for both vaccines and drugs. The spike is exposed on the surface of the virus, where it waits to latch onto a host cell and initiate infection. Because of its accessibility and crucial role in the viral replication cycle, the spike has been the focus of the vaccine development efforts and a major target for drugs and antibodies. It also caught our attention because it must undergo significant structural changes to perform its function, but it has been impossible to directly observe these motions. At least until our simulations captured them! This amazing computational feat has revealed new ways to target the spike, explained ‘cryptic’ antibody binding sites that appeared to be inaccessible in available experimental snapshots of the spike, and shed light into how changing the spike changes the pathogenicity of the virus. Now we are building on this work to understand how emerging versions of the spike differ from the original SARS-CoV-2 virus and how we can attack them.
We also searched the SARS-CoV-2 proteome for ‘cryptic’ pockets that may provide new opportunities for drug design. Often snapshots of proteins derived from experiments only reveal limited opportunities for drug design. In many settings, we’ve found that watching the dynamics of these proteins reveals the spontaneous formation of pockets that were absent in the experimental structures, which we call cryptic pockets. So far, we have found over 50 of these cryptic pockets in proteins from the SARS-CoV-2 virus. A pocket in one protein, called Nsp16, is particularly appealing for drug design because we also found that it is present in the SARS-1 and MERS viruses, so a drug that targets this pocket might work against all coronaviruses related to SARS-CoV-2.
In a third major effort, we joined forces with the COVID Moonshot to develop a patent-free antiviral that targets a viral protein called the main protease. This collaboration brings together a variety of computational and experimental research teams from across the globe. So far, we have found multiple lead compounds that show efficacy in experimental tests, and the project is now performing the first tests in animal models. You can read more here.
You can learn more about our COVID-19 work here.
In addition to all the scientific advances, we welcomed a number of new members to the Folding@home consortium. Diwakar Shukla (UIUC) and Xuhui Huang (HKUSST), both of whom are Pande lab alum, rejoined the Folding@home effort. Lucie Delemotte (KTH) and Erik Lindahl (Stockholm University) both joined our community. And we welcomed Emma Matthies as a program manager.
We are also grateful to many new friends that have supported our efforts. Thanks to Avast, AWS, Cisco, Linus Tech Tips, Microsoft Azure, Oracle, and VMware for helping us to scale-up Folding@home’s server-side infrastructure to keep up with the tremendous growth we experienced in such a short time. Thanks to Microsoft AI for Health for helping us use Azure to run adaptive sampling simulations, and to UKRI for providing compute resources to parallelize data analysis. Thanks to Pure Storage for providing a FlashBlade system to store our large datasets, to Seagate and Micron for additional storage, and to MolSSI for helping organize public datasets. Thanks to AMD, ARM and Neocortix, and Intel for helping to improve the performance of Folding@home on their hardware. Thanks to all of these companies for helping to spread the word about Folding@home, and also to A16Z, Best Buy, CCP, CoreWeave, Daimler Truck AG, Dell, GitHub, HP, La Liga, Media Monks, Microcenter, NVIDIA, and Telefonica. Thanks to CERN and the particle physics community for helping with data management and to DataDog for server monitoring services. We are also extremely grateful to the NSF and NIH for funding.
While we are happy to see 2020 in the rearview mirror, one silver lining of the year was the sort of family reunion we had as people sought to help with Folding@home and our COVID-19 work. Many people have participated in Folding@home over the past 20 years, and many redoubled their efforts to help the project as we collectively sought to drive progress on SARS-CoV-2 as quickly as possible. Thanks to all of you!
Finally, we are extremely grateful to everyone who contributed their compute power to Folding@home! Without you, we couldn’t have achieved so much.
We look forward to continuing to work with you, and wish you a happy new year!