As we all know, coronaviruses are a major threat to human health. In the past two decades these viruses have caused three epidemics: SARS in 2004, MERS in 2012, and now, COVID-19 has caused a global pandemic. The painfully obvious questions to address are 1) how do we prevent coronavirus-caused diseases? and 2) how do we treat people who get sick with these diseases? Fortunately, the newly approved vaccines are a great step on question 1. However, the second question is an open question.
Like all viruses, coronaviruses employ molecular machines, called proteins, to carry out a cycle of infecting host (human) cells and replicating. If you can disable the function of these proteins, the viruses can’t continue this cycle! Therefore, figuring out how these proteins operate and devising ways to “turn off” these proteins is a core focus when developing a drug meant to treat someone who is infected.
In our most recent work, we use computer simulations to understand the moving parts of an essential SARS-CoV-2 protein. While we can usually figure out the general shape (i.e. 3D structure) of a protein with various experiments (including shooting x-rays at the protein), even the most powerful microscopes in the world cannot show us how these proteins move! Computer simulations show us how a protein actually moves, which can both inform us how they function and show us new ways to disable them.
The coronavirus protein we study in this work is called Nsp16. It helps the virus evade our immune response, so if we could disable Nsp16, then our immune system would more easily recognize the virus and knock it out. People have previously tried to disable Nsp16 by wedging a drug into its “active site” (i.e. the region where it performs a reaction to disguise viral RNA from the human immune system). In the case of Nsp16, the active site consists of two pockets, one that binds RNA and one that binds another molecule called SAM. However, there is a similar human protein (CMTr1) that has a similar looking active site, so targeting the coronavirus protein without disabling the human protein is challenging.
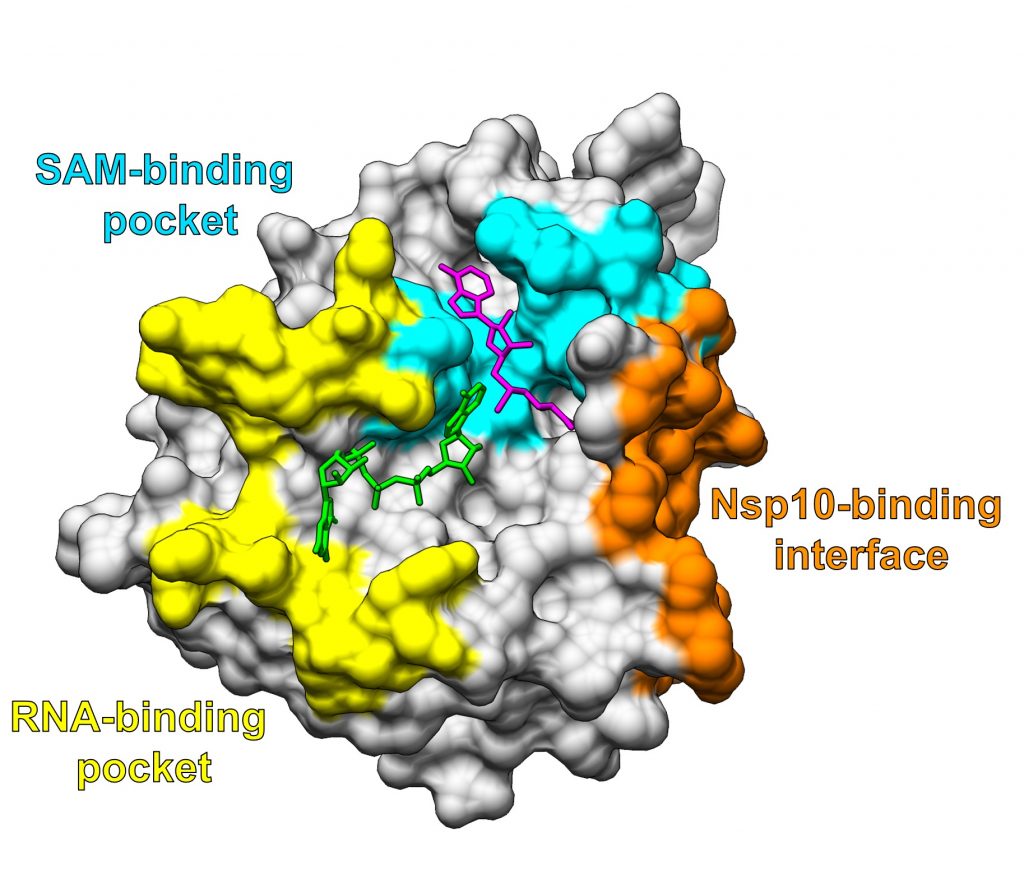
One important difference distinguishing human relatives of Nsp16 from the coronavirus Nsp16 protein is that the coronavirus protein needs to be attached to another viral protein, Nsp10, to carry out its function.
In this work, we first show how Nsp10 binding to Nsp16 activates Nsp16, and then, by comparing motions of Nsp16 and its human homolog (CMTr1) we identify a way to disable Nsp16 without disabling the human protein.
First, we showed how Nsp10 binding to Nsp16 activates Nsp16 to be able to function. Specifically, when Nsp10 attaches to Nsp16, Nsp16’s 3D structure changes so that its active site is more open (shown in the movie below). With a more open active site Nsp16 is able to bind to RNA and SAM, which are both required to enable the RNA-disguising reaction!
Next, we find a way to target coronavirus Nsp16 without targeting an important human relative, the protein CMTr1. Specifically, we find that some of Nsp16’s motions lead to the formation of a ‘cryptic’ pocket in a region of the protein near the active site, but human CMTr1 does not form this pocket! Importantly, when this pocket is open, the active site is closed, meaning Nsp16 likely cannot bind to the necessary molecules that it needs to disguise its genome! Therefore, if someone can develop a small molecule to stick into and wedge open this pocket, it would likely disable the protein. And, we find that Nsp16 proteins of other coronaviruses also have this pocket, so targeting this pocket could knockout other coronaviruses (like SARS, MERS, etc.) Fortunately, this would likely not affect human CMTr1 since it does not form this pocket.