Written by: Ivy Zhang, Viktor Belay, William Glass
The Chodera lab is investigating a new class of SARS-CoV-2 antibodies currently in clinical trials that works through a surprising new mechanism that just may suppress the ability of the virus to evolve resistance.
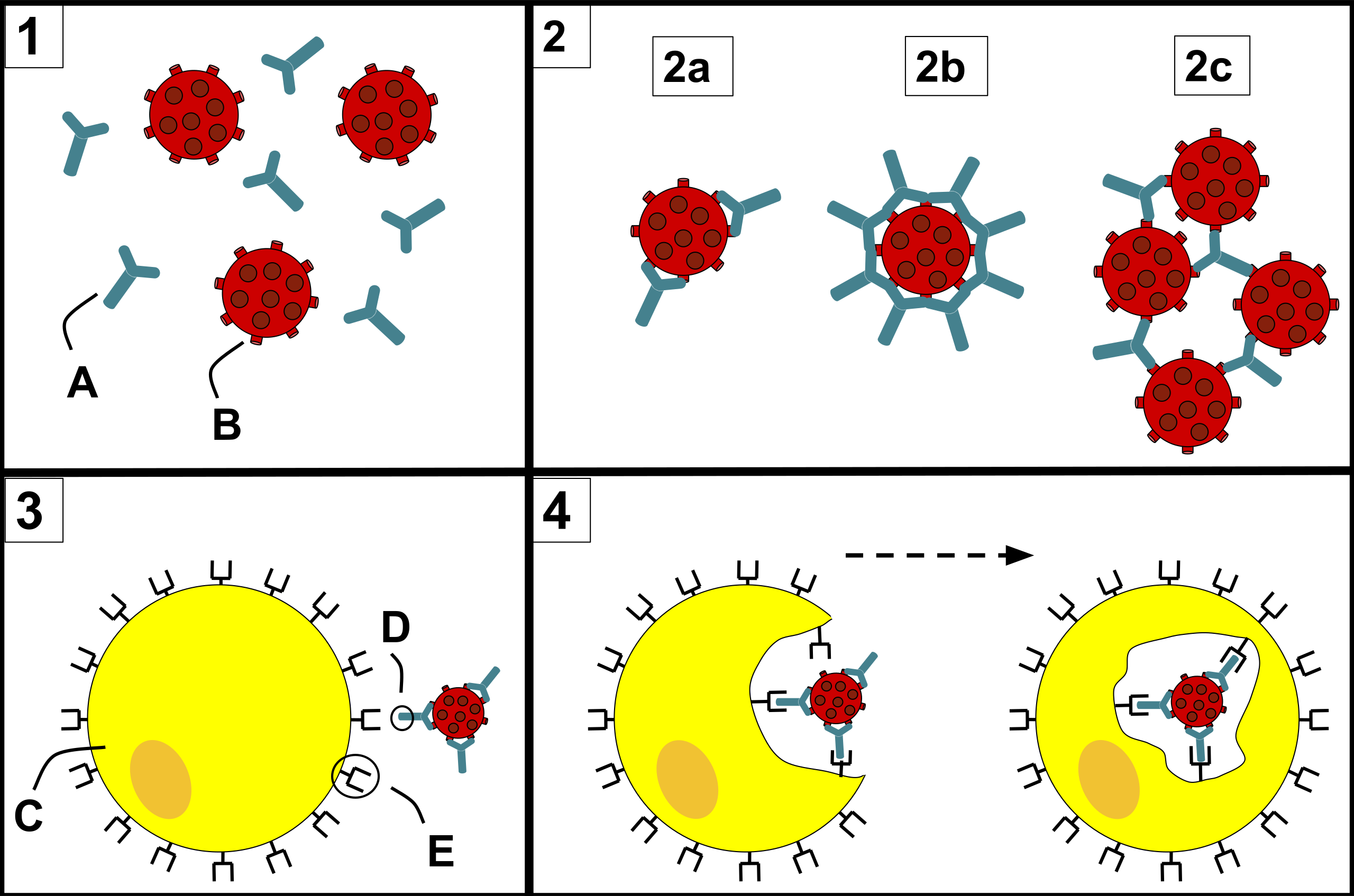
Antibodies are one type of drug being developed for treatment of COVID-19
Antibodies are proteins generated by our immune systems to protect us from infectious diseases, like COVID-19. At a high level, all antibodies prevent infection in a similar manner: they bind to proteins on the disease-causing agent (e.g. virus), interfering with the disease-causing agent’s ability to enter human cells. For our immune systems, SARS-CoV-2, the virus causing COVID-19, is no exception. Our immune systems generate antibodies to neutralize SARS-CoV-2, meaning they prevent the virus from entering our cells and infecting us. One important caveat is that this natural form of defense is most effective in young, healthy individuals; older individuals and those with pre-existing conditions that render their immune systems weaker than normal may not be able to generate these antibodies as effectively to battle COVID-19.
{kind=link}
One major avenue for COVID-19 drug development involves trying to reproduce our bodies’ natural immune response. A number of pharmaceutical and biotech companies have been developing artificially-produced antibodies to serve as drugs to both treat COVID-19 and provide some temporary (weeks to months) protection against SARS-CoV-2 infection. There are many antibodies in clinical trials right now, most of which have a pretty obvious mechanism of action (manner in which they neutralize the virus): directly blocking the virus from binding to human cells. However, some of the antibodies in clinical trials do not have an easily explained mechanism of action, as they do not directly block viral entry. Intriguingly, they bind to a conserved region on the virus that may not be able to easily mutate, meaning these antibodies may still be effective even as the virus mutates.
As mentioned above, these antibodies that are currently under development aim to protect against SARS-CoV-2 infection by disrupting viral entry, but moving to a deeper level of how the things work: what do and don’t we know about how these antibodies neutralize SARS-CoV-2?
What we know: The RBD is a common target for SARS-CoV-2 antibody development
For SARS-CoV-2, viral entry is dually mediated by the spike protein (spike) within the viral membrane and angiotensin-converting enzyme 2 (ACE2) within the human cell membrane. Both the spike and ACE2 are known as glycoproteins, which are proteins that contain long molecules called glycans. The effect of glycans on biomolecular pathways are numerous and we still don’t fully understand every process they are involved in. However, in SARS-CoV-2 they have been shown to be instrumental in how the spike functions [1].
The part of the spike that binds to its receptor, ACE2, is called the receptor binding domain (RBD). To mitigate SARS-CoV-2 infection, many artificially-produced antibodies compete with ACE2 for RBD binding, directly blocking the ACE2:RBD binding event required for viral entry.
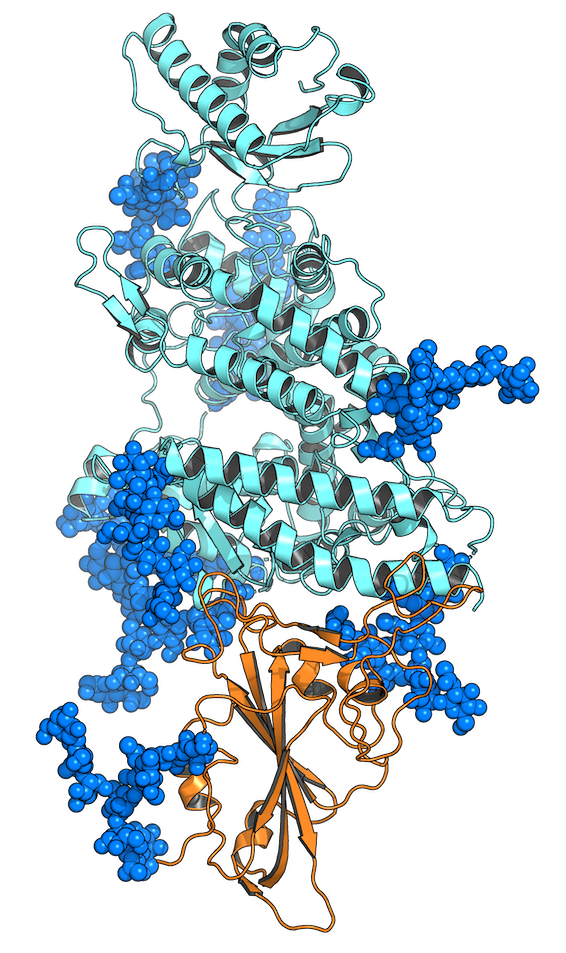
What we don’t know: Pinto et al. discover S309, a neutralizing antibody with an unknown mechanism of action
Some studies have reported virus-neutralizing antibodies that bind to a different side of the RBD from ACE2, preventing infection through an unknown mechanism. (For a summary of the various binding modes of antibodies currently under development, see Barnes et al. [2])
In “Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody”, Pinto et al. demonstrate that several antibodies derived from a patient that survived SARS-CoV in 2003 are able to neutralize SARS-CoV-2 [3]. They found that one antibody is particularly effective at neutralization: S309, which is currently in clinical trials. However, cryo-EM images of the RBD:S309 structure reveal that S309 does not function as many other SARS-CoV-2 antibodies do; rather than competing with ACE2 for RBD binding, S309 binds to a different side of the RBD than ACE2 binds. In fact, the RBD still binds to ACE2, but the virus is somehow still neutralized.
Moreover, the S309 binding site on the RBD is more conserved than the site where ACE2 binds, meaning that as the virus mutates, the S309 binding site will be less likely to mutate than other regions of the RBD. This suggests that S309 may be resistant to viral mutations, meaning that it could continue to bind and neutralize SARS-CoV-2 even as it evolves.
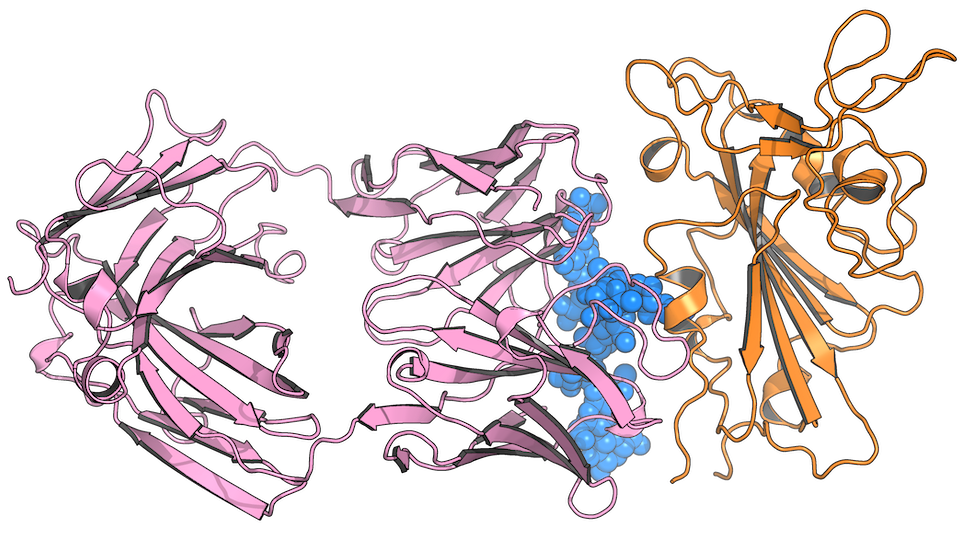
So how does S309 neutralize SARS-CoV-2? This is still an open question and we are interested in answering it because of S309’s potential resistance to viral mutations. Any new knowledge we can uncover related to S309’s mechanism of action will be useful for comparing it to other SARS-CoV-2 antibodies under development, which may help inform doctors on the best treatment to prescribe to patients with different mutational variants of SARS-CoV-2. Furthermore, building a better description of how S309 interacts with the RBD will enhance our understanding of the SARS-CoV-2 infection process, which will put us in a better position to combat future outbreaks of SARS-CoV-2-related viruses, should they emerge.
We will use Folding@home to investigate S309’s mechanism of action
While the structural data presented by Pinto et al. [3] reveals some key insights into how the S309 interacts with the RBD, it only provides a static snapshot of the RBD:S309 complex. In reality, proteins are dynamic and constantly in motion.
The Chodera lab runs molecular dynamics (MD) simulations (using OpenMM) to study the motion of proteins over time. However, MD simulations are computationally expensive, so if we want to study protein motion over a long timescale, we need a lot of computational resources. This is where Folding@home comes in! With the enormous aggregate computing power of Folding@home, we can generate simulation data at unprecedented scales. We can then use the simulation data to make detailed movies of the proteins in motion and more importantly, gain insight into the conformational states that a protein of interest can adopt.
Why do we care about the conformational states of a protein? Each protein adopts a range of conformations, where each conformation dictates the protein’s interactions with its binding partners, thereby influencing the protein’s function. Furthermore, the range of conformational states can change in the presence of a new binding partner.
We can apply these ideas to the RBD and try to understand S309’s mechanism of action from the perspective of protein conformational change. We hypothesize that when S309 binds to the RBD, it alters the set of conformational states of the RBD such that the RBD no longer binds to all the same partners, disrupting the virus’s ability to enter human cells. To investigate this hypothesis, we will run simulations of apo RBD (Project 17307), apo ACE2 (Project 17308), RBD:ACE2 (Project 17309), and RBD:S309 (preparation for F@h in progress) and compare the range of conformational states for the RBD across these simulations.
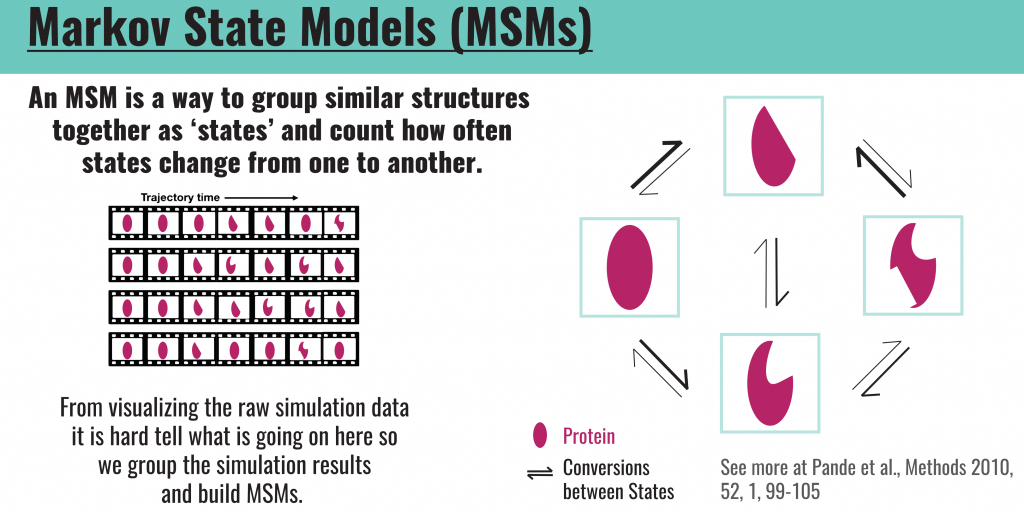
Each simulation will output a set of all of the conformations that the structure was in during the time period of the simulation. Each conformation can correspond to the movement of a single atom within the protein structure. Considering that proteins can be composed of thousands of atoms, the simulation output sets will be extremely large. In order to make sense of these huge volumes of data, we build Markov State Models (MSMs), which are frameworks for analyzing dynamic systems, such as proteins. MSMs allow us to group similar conformations as “states.” Once the data is organized in this way, we can determine how the conformational states of the RBD change upon S309 binding and whether these observations help explain S309’s mechanism of action.
Stay tuned for updates on our progress!
References:
[1] Casalino L., Gaieb Z., Goldsmith, J.A., et al. Beyond shielding: the role of glycans in the SARS-CoV-2 spike protein. ACS Central Science (2020). https://doi.org/10.1021/acscentsci.0c01056
[2] Barnes, C.O., Jette, C.A., Abernathy, M.E. et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature (2020). https://doi.org/10.1038/s41586-020-2852-1
[3] Pinto, D., Park, Y., Beltramello, M. et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature (2020). https://doi.org/10.1038/s41586-020-2349-y