The spike of the SARS-CoV-2 virus (shown below) is one particularly appealing target for designing therapeutics to combat the COVID-19 disease. It is actually comprised of three identical proteins arranged in a circle. Many copies of the spike protrude from the surface of the virus, where they wait to encounter a protein on the surface of many human cells, called ACE2. Binding of a spike to ACE2 initiates a series of events that ultimately allow the virus to enter the human cell. Therefore, therapeutics that bind the spike in a manner that blocks its interaction with ACE2 could provide a valuable means to prevent infection.
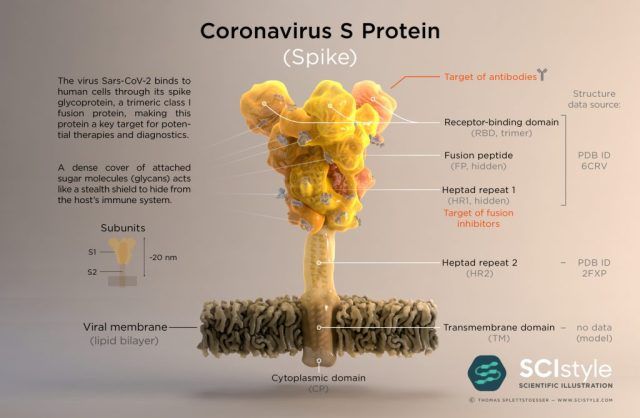
While extremely valuable, the structure shown above is far from a complete picture of how the spike works, or the ways that we could target it with therapeutics.
In fact, it is well-established that the spike must undergo a dramatic opening motion to reveal the interface that ultimately binds a human cell. Specifically, the spike has three receptor-binding domains that directly bind to ACE2 (the protein on the surface of human cells). For the spike-ACE2 interaction to form, the spike’s three receptor-binding domains must open up to reveal the binding interface.
The opening of the three receptor-binding domains reminded us of the mouth of the Demogorgon monster from the television series Stranger Things, so we’ve taken to calling the spike the COVID-19 Demogorgon.
Understanding how the spike opens up to engage the ACE2 protein could be extremely useful. Every step along the way could potentially be targeted with therapeutics.
Unfortunately, there is no way to watch a spike undergo this transition, at least with existing experimental techniques. Data on what the open state even looks like is also limited.
To overcome this limitation, we are using computer simulations to watch the COVID-19 Demogorgon open its mouth. To do this, we are using a two-stage process. In the first stage, we use an algorithm called FAST adaptive sampling that allows us to focus simulations on capturing specific structural changes, like the opening of the spike. While we can see these processes, it’s often still hard to say how long each stage of the process takes. Such information is valuable because long-lived steps are generally better therapeutic targets. To get that information, we then take representative structures from different stages of the opening process and start thousands of simulations on the Folding@home distributed computing platform. These simulations let us identify additional structures of the protein, determine the relative lifetimes of all the different structures, and prioritize them for drug development.
Both steps are extremely computationally expensive. If you tried to simulate the opening of the spike on your home computer, you’d be lucky to see even part of the process within the next 100 years. Even with the reasonably powerful computer cluster we run at the Washington University School of Medicine, we’d be hard put to capture this event. Needless to say, none of us wants to wait that long for a COVID-19 treatment to be developed.
Fortunately, we have reinforcements! Over the past three weeks, over 700K citizen scientists have joined the Folding@home project. This 20-fold increase in participation has given us unprecedented compute power to bring to bear on COVID-19. In fact, Folding@home is the first computer to achieve the exaFLOPS scale and is now estimated to be more powerful than the next top 100 supercomputers combined.
We have also established a number of collaborations with industry. For example, John Kahan, Chief Data Analytics Officer and AI for Health lead at Microsoft, shares our vision, saying, “The scale and size of the current global pandemic requires a collaborative approach between companies, non-profits, governments, scientists, and clinicians. We are glad to be able to support the Folding@home project through Washington University School of Medicine in the acceleration of COVID-19 related research using Azure cloud resources.” By joining forces with the AI for Health and Azure teams at Microsoft, we’ve been able to bring significantly more compute power to bear on the first stage of our simulations. They’re also helping us host additional servers to better serve the Folding@home community.
With our powers combined, we’ve captured the opening of one of the receptor-binding domains of the COVID-19 Demogorgon, shown below.
Partial opening of the mouth of the COVID-19 Demogorgon (aka spike) captured by our simulations. The three colors are the three proteins that come together to form the spike. Each is made of a linear chain of chemicals called amino acids. The ribbons trace out each chain. The transparent surface is the surface of the COVID-19 Demogorgon. The three proteins that make up the Demogorgon must spread apart to reveal the ACE2 binding site, which initiates infection by attaching to a protein called ACE2 on the surface of human cells. This movie captures part of that opening motion.
Thanks to our great community for all the help! We couldn’t have done this without you.
We’ll be continuing to run additional simulations to capture the opening of the other receptor-binding domains. Once the data are ready, we’ll be making it publicly available.
Of course, please bear in mind that this work is at the bleeding edge of science. We’re throwing everything we’ve got at COVID-19, but we can’t say for sure if/when our work will lead to treatments.
With that said, you can help us maximize the chances for success!
- Downloading Folding@home and helping us run simulations is the primary way to contribute. These calculations are enormous, and every little bit helps! Each simulation you run is like buying a lottery ticket. The more tickets we buy, the better our chances of hitting the jackpot. Usually, your computer will never be idle, but we’ve had such an enthusiastic response to our COVID-19 work that you will see some intermittent downtime as we sprint to setup more simulations. Please be patient with us! There is a lot of valuable science to be done, and we’re getting it running as quickly as we can.
- If you don’t have computers to contribute or are feeling particularly generous, you can also make donations through Washington University in St. Louis. These funds are used for a number of purposes, including: 1) supporting our software engineering and server-side hardware (particularly important right now as we scale up rapidly!) and 2) buying compounds to test experimentally based on insight from our simulations.