TL;DR: Today, we are excited to share two papers that tell distinct stories about AI drug discovery:
1. “Spatial Graph Convolutions for Drug Discovery” describes new deep neural network architectures for modeling drug-receptor interactions. We argue that the future of predicting the interactions between a drug and its prospective target demands more than simply applying deep learning algorithms from other domains, like vision and natural language, to molecules. https://arxiv.org/abs/1803.04465
2. “Machine Learning Harnesses Molecular Dynamics to Discover New μ Opioid Chemotypes” describes an algorithm that leverages protein motion to enrich the search for active molecules. We then applied the method to find a new chemical scaffold that we experimentally verified is an agonist for the µ Opioid Receptor. https://arxiv.org/abs/1803.04479
Introduction
From the era of the hunter-gatherers to the present, starting long before the discovery of atoms, humans have treated disease by searching for small organic molecules with therapeutic properties. From anti-inflammatory tree barks in the bronze age to COX-2 selective inhibitors in the information age, the vast majority of drugs are small molecules that bind to one or several biological macromolecules. The advent of chemistry and atomic theory promised to catalyze drug discovery through rational design. Through X-ray crystallography, high resolution structures of target proteins could be solved, empowering medicinal chemists — at least in theory — to design molecules with high specificity for drug targets.
While the field of drug discovery has made strides in rational design, a relatively small portion of today’s drug discovery pipeline is conducted in silico. Novel lead molecules are mostly found through experimental high-throughput virtual screens; lead optimization is mostly pursued through often painstaking structure-activity relationship development and synthesis of derivatives based on the intuition of the chemist; and toxicity prediction is mostly done through in vitro and in vivo assays. With minimal use of computers, a concert of academia, government, and corporate pharmaceutical laboratories have developed an effective pharmacopeia capable of reducing pain, treating many cancers, and mitigating heart disease. In light of this success, one might reasonably inquire, why devote so much effort to integrating computational chemistry into the pipeline of drug discovery?
Experiments, while accurate, are expensive. Time and money are precious resources when the vast majority of compounds fail to reach FDA approval and those that do cost $1.2 billion on average to research and develop. When searching for lead molecules, it costs about $100 to purchase a single compound in a commercially available library; in the lead optimization phase, it costs about $2500 to synthesize a proposed derivative; up to another $2500 for functional assays of candidate ligands; and the subsequent mouse-model and human studies that follow a successful lead optimization campaign cost exponentially more. A simple back-of-the-envelope calculation shows that experimentally testing all 100 million purchasable compounds in the ZINC small molecule database is financially intractable for even the best funded laboratories. Even then, the ZINC database is a small portion of the vast combinatorial expanse that is drug-like chemical space.
In contrast, computational models are on average much faster and less expensive than their experimental counterparts. Thus far, the utility of computational chemistry in drug discovery has been limited by its comparative inaccuracy. There are several sources of this inaccuracy. Since quantum methods are computationally infeasible on the scale of protein-ligand interactions, a Newtonian approximation is usually made. As a result, techniques like molecular dynamics — which simulate the conformational changes of a macromolecule — and molecular docking, which estimates the binding affinity between a protein and ligand, are intrinsically limited by force field error. Compounding this error, drug discovery scientists typically dock to single crystal structures of target proteins even though those proteins sample many conformationally distinct and biologically relevant states.
In this family of works, we seek to address several of these limitations to computational drug discovery by synthesizing existing methods in new ways and developing new fundamental technologies.
Paper 1: Spatial Graph Convolutions for Drug Discovery
Can artificial intelligence (AI) resolve such vexing tradeoffs? While machine learning (ML) has made immense progress in the fields of computer vision and natural language processing, it has yet to offer comparable improvements over domain-expertise driven algorithms in the molecular sciences. One can attribute this achievement gap to two shortcomings: (1) ImageNet contain over ten million labeled examples whereas the PDBBind and Tox21 datasets have less than ten thousand examples; (2) current neural network architectures for molecules are in their infancy compared to the advanced convolutional and recurrent counterparts for vision and NLP.
In the first paper we describe novel deep learning algorithms specifically designed to model protein-ligand complexes. Both new deep neural network architectures naturally encode the symmetries inherent in drug-receptor systems, which include both bonded and non-bonded interactions. On the gold standard benchmark of the field — the PDBBind dataset — we achieve state-of-the-art results for predicting the affinity with which a given ligand binds to a protein target of interest. Rapidly, inexpensively, and accurately predicting protein-ligand binding affinity empowers pharmaceutical scientists to find and optimize molecules to enact specific therapeutic outcomes. Specifically, it enables one to design molecules with maximal affinity for a desired target, minimal affinity for undesired targets, and superior metabolic properties.
How does our method work? Let’s begin with the graph convolution. The most successful DNN architectures are constructed to exploit the unique structure of the data at hand. For instance, convolutional neural networks (CNNs) leverage the information encoded in proximity between pixels in images, whereas recurrent neural networks balance the significance of both neighboring and separated blocks of text in modeling natural language. In approaching the problem of protein-ligand binding affinity prediction, one might be tempted to draw an analogy to computer vision problems. Just as neighboring pixels connote closeness between physical objects, one could subdivide a binding pocket into a grid of voxels, where neighboring voxels denote neighboring atoms and blocks of empty space. Unfortunately, such a 3D CNN approach has several potential drawbacks. First, it is highly memory intensive in both the inputs and in the hidden weights. Second, the parameters, growing exponentially as the number of dimensions, suffer from a “curse of dimensionality”.
By treating atoms as nodes and bonds as edges, graph convolutions can be viewed as a hierarchical, differentiable, and flexible analogy to the commonly used circular fingerprint representation in cheminformatics. For those familiar with molecular dynamics simulations, a graph convolution can be seen as generating differentiable atom types. To represent a carbon bonded to four other atoms, for example, a 3D CNN will need several different filters to accommodate the many symmetrically equivalent orientations that such a functional subgroup can undertake. In contrast, a graph convolution is symmetric to permutations and relative location of each of the four neighboring atoms, thereby saving a sizable number of parameters. The basic update of a graph convolution (right) can be viewed by analogy to a fully connected neural network (left):
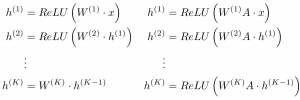
Here, A is an N by N square adjacency matrix (a “1” in row i, column jrepresents a bond between atoms i and j), x is an N by p matrix of input “features” for each atom, ReLU is a type of pointwise nonlinearity, and W is a weight matrix. Illustrated visually:
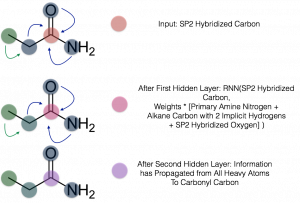
Figure 1: Schema of graph convolutions for the carbonyl carbon of propanamide
So, how does one generalize this powerful concept to tackle drug-protein binding complexes? To those coming from a pure computer science background, one can view a drug-protein complex as two interacting graphs, where the atoms and bonds of the drug graph are in some spatial proximity to the atoms and bonds of the protein graph, as shown in Figure 2.
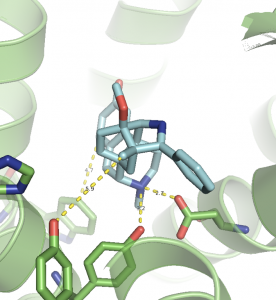
Figure 2: Morphine-like molecule interacting with µ Opioid Receptor
By generalizing the definition of adjacency to include spatial proximity in addition to bonds, we can define such a complex as a single, large graph, where nodes are still atoms but edges can now be of multiple different types. In mathematical terms, this would be tantamount to expanding the N by N adjacency matrix A introduced above to an N by N by Nedge-types adjacency tensor. In one Spatial Graph Convolution, which we call the Staged Spatial Graph Convolution, we stack end-to-end two graph convolutional and one fully connected network; the first graph convolution is only over bonded edges and can be viewed as deriving differentiable atom types or fingerprints; the second graph convolution incorporates spatial neighborhoods between the newly derived atom types; and the final fully connected layers compute a binding free energy:
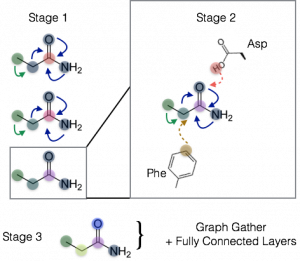
Figure 3: Illustration of staged spatial graph convolution
We also introduce a related spatial graph convolution which we term, “PotentialNet,” which synthesizes the bond- and distance-based graph convolutions in a single stage. Here, the network decides how to perform a graph convolutional atom type update by integrating information about both the neighboring atoms and the type of edge that connects them.
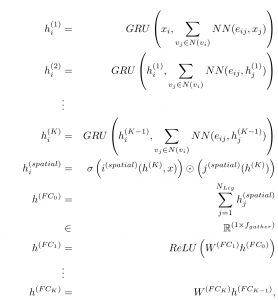
Figure 4: Mathematical description of PotentialNet Spatial Graph Convolution
Here, inspired in part by Gated Graph Neural Networks and Message Passing Neural Networks [1, 2], we use the Gated Recurrent Unit (GRU) operation. Intuitively, at each graph convolutional step, for each atom, the GRU operation decides which features of that atom to “forget” and then adds only those features from neighboring atoms (whether bonded or close in real space) that are relevant to binding energy prediction.
Paper 2: Machine Learning Harnesses Molecular Dynamics to Discover New μ Opioid Chemotypes
Developing fundamentally new molecules demands creative approaches to their discovery. For decades, researchers have used three key methods for drug discovery: structural biology (e.g. crystallography), molecular simulation (e.g. molecular dynamics, docking), and machine learning (e.g. QSAR, Random Forests). However, these methods have been used independently, as there has been no scheme that can synthesize these seemingly orthogonal methods. In this work, we present a novel algorithm drawing on all of these approaches in order to greatly improve our predictive ability in drug design. The success of this scheme supports a key hypothesis in protein function more broadly: protein receptors sample a complex conformational landscape in their functional duties.
Chemists typically assay drug candidates by virtually screening compounds against crystal structures of a protein despite the fact that they traverse many non-crystallographic states. This multiplicity of states is inaccessible to current drug discovery scientists. The µ Opioid Receptor (µOR) — the primary target for clinically-used opioid analgesics — exemplifies such conformational diversity. In this work, using unprecedented millisecond-scale molecular dynamic simulation, we first discover new functionally relevant conformational states of µOR. Subsequently, we machine learn ligand–structure relationships to substantially improve prediction of opioid ligand function. Incorporating non-crystallographic states of GPCRs therefore enhances the enrichment of active compounds.
We use unsupervised machine learning (ML), including the time-structure based independent component analysis (tICA) algorithm as well as K-Means clustering. The tICA algorithm was applied to reduce the dimensionality of the MD simulation data, an operation that facilitated both the analysis of the slow dynamical modes of µOR as well as the fidelity of the clustering into discrete conformational states of the receptor. Subsequent to this unsupervised step, we dock a library of drugs with known efficacies (agonism vs. antagonism) and affinity for µOR to each of the two available crystal structures of the receptor as well as a set of new conformations derived from K-Means and tICA analysis of MD simulation. Finally, in the supervised ML step, we build random forest classifier models that map the set of (ligand_i, conformation_j) docking scores to binarized labels for affinity and agonism. We report substantial increases in enrichment, as measured by Receiver Operating Characteristic Area Under the Curve (AUC), by incorporating the MD-derived conformers in addition to the crystal structures.
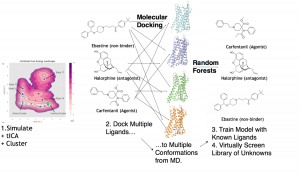
Illustration of technique for exploiting molecular dynamics with machine learning for activity prediction
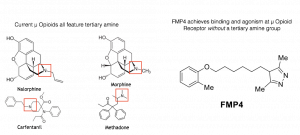
Why focus on µOR? In a dispiriting trend, more than 30,000 Americans died of opioid drug overdose in the year 2015, up from 20,000 only five years earlier. Over the past century, medicinal chemists have striven in vain to synthesize an opioid analgesic without issues of dependence. A significant portion of currently known opioids are analogs of one of two scaffolds: morphine and fentanyl. The vast majority of currently known opioids are centered on a tertiary amine nitrogen motif. Departing from this century-old strategy of creating derivatives, we seek to open completely new regions of chemical space for development of a novel class of µ opioids.
In collaboration with Susruta Majumdar and Gavril Pasternak of Memorial Sloan-Kettering Cancer Center in New York, we strove to plant our flag in such uncharted opiate chemical territory. We virtually screened over one hundred thousand molecules from the Stanford Compound Library. After testing the top thirty scoring compounds in their laboratory, Majumdar, Pasternak, and colleagues were pleased to report that we had discovered a novel chemotype active at the µ Opioid Receptor. One molecule, which we called FMP4, has micromolar affinity for µOR and is an agonist for the receptor as well. By measure of the Tanimoto score, which assesses chemical similarity on a scale of 0.0 to 1.0, FMP4 has a maximum Tanimoto score of 0.44 to any currently previously known opioid.
Closing Remarks
The first use of the term “rational drug discovery,” according to Google Scholar’s records, may be from Richard D. Cramer, the prominent computational chemist best known for his work with Tripos, Inc. The program of identifying a promising macromolecular target, resolving to atomic resolution a binding pocket of that target, and finally engineering molecules to bind to and modulate that target for therapeutic effect is as compelling and inspiring an idea today in 2018 as it was in 1982. If any conclusion can be drawn from these past four decades of tremendous effort in “rational drug discovery,” it is that there is unlikely to be a single magic bullet that will be a generic technique for generating new medicines.
In light of this recalcitrant complexity, one must caution against the continuing divide between experimental and computational scientists in the common fight against disease. Deep learning may be reaching a climactic phase of the hype cycle, but it would have no utility in computer-aided drug discovery were it not for the tireless effort of force field developers and crystallographers in heightening our understanding of drug-target interactions. We in the Pande Lab believe that, in the next four decades, it will be those brave or bold enough to reach across the street and develop methods at the interface between fields that will make the most progress in realizing the vision of computer-aided drug discovery.
References
[1] https://arxiv.org/pdf/1511.05493.pdf
[2] https://arxiv.org/pdf/1704.01212.pdf
Post by Pande Lab member Evan N. Feinberg