The so-called “guardian of the genome”, p53 is a tumor suppression protein involved in regulating cell repair and apoptosis. Many cancer cells are able to proliferate because they have mutations to p53, making it the focus of intense interest for cancer researchers.
In the cell, levels of p53 are usually kept low through the action of another protein, MDM2, which binds to the transactivation domain (TAD) of p53 and recruits it for degradation. Upon binding, the p53 TAD folds into a helix and packs into a binding cleft of MDM2. Over the last ten years, there have been many efforts to develop cancer-fighting molecules that can mimic the p53 helix and competitively bind MDM2, resulting in the up-regulation of p53.
MDM2 is interesting for another reason: NMR experiments show that in the absence of p53, the disordered N-terminal region becomes partially structured and associates with the cleft. This so-called “lid region” can be seen making key interactions with MDM2 ligands in co-crystal structures, which suggest that the ability to predict likely lid conformations might be very important to rational drug design.
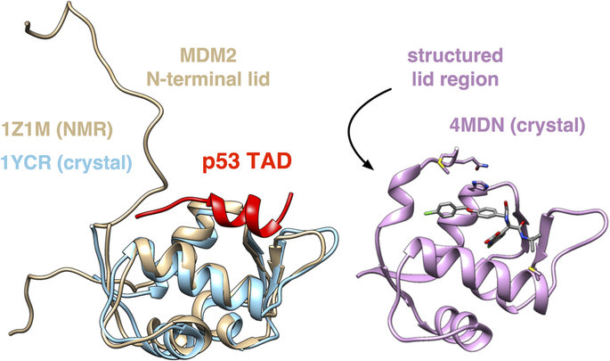
In a new paper in Scientific Reports, researchers from the Voelz lab have applied the power of distributed computing through Folding@home to study the conformational dynamics of the MDM2 lid region, to discover just how important modeling the lid region may be. Markov State Models (MSMs) built from simulated trajectories of the lid region show that the lid associates with the binding cleft in a two-state manner consistent with experiment, and furthermore that this motion is coupled to the opening up of the binding cleft. Since the published NMR structure of MDM2 has a closed cleft, the researchers then went on to see if known ligands could be successfully docked into the simulated MDM2 receptor structures. Remarkably, this procedure was highly successful, comparable to the “gold standard” of cross-docking ligands across a set of high-resolution co-crystal structures.
These results have important implications. For one, they suggest that large-scale simulations can help refine “bad” receptor structures for the purpose drug design. This might be particularly important for homology models of protein structures, for which computational docking often fails. The results also suggest that modeling disordered regions in proteins might be more important that previously thought.
Reference:
Mukherjee, S., Pantelopulos, G. A., & Voelz, V. A. (2016). Markov models of the apo-MDM2 lid region reveal diffuse yet two-state binding dynamics and receptor poses for computational docking. Scientific Reports, 1–10. doi:10.1038/srep31631