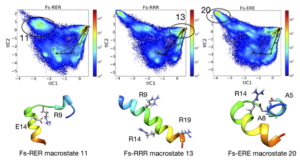
Salt−bridges are electrostatic interactions between positively charged residue side chains (like arginine or lysine) and negatively-charged side chains (like aspartic and glutamic acids). They play an important role in stabilizing many protein structures, and have been shown to be designable features for protein design.
In this work, we study the effects of non-native salt bridges on the folding of a soluble alanine-based peptide helix (Fs peptide) using extensive all-atom molecular dynamics simulations performed on the Folding@home distributed computing platform. Using Markov State Models, we show how non-native salt-bridges affect the folding kinetics of Fs peptide by perturbing specific conformational states. Furthermore, we present methods for the automatic detection and analysis of such states.
Why are these results important to our understanding of protein folding? It used to be thought that protein unfolded states resembled a “random coil” with few structural features. We now know that salt-bridges forming in unfolded states can significantly affect folding stability and dynamics. Our work shows that we can quantitatively predict these effects. Eventually, we hope to be able to use such knowledge to engineer the folding properties of proteins.
Just Accepted manuscript: http://dx.doi.org/10.1021/acs.jpcb.5b11767