V. Voelz, V. Singh, W. J. Wedemeyer, L. Lapidus, and V. S. Pande.
Journal of the ACS, 132 4702–4709 (2010)
ABSTRACT.
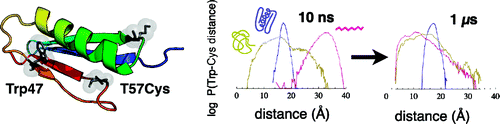
While several experimental techniques now exist for characterizing protein unfolded states, all-atom simulation of unfolded states has been challenging due to the long time scales and conformational sampling required. We address this problem by using a combination of accelerated calculations on graphics processor units and distributed computing to simulate tens of thousands of molecular dynamics trajectories each up to 10 μs (for a total aggregate simulation time of 127 ms). We used this approach in conjunction with Trp-Cys contact quenching experiments to characterize the unfolded structure and dynamics of protein L. We employed a polymer theory method to make quantitative comparisons between high-temperature simulated and chemically denatured experimental ensembles and find that reaction-limited quenching rates calculated from simulation agree remarkably well with experiment. In both experiment and simulation, we find that unfolded-state intramolecular diffusion rates are very slow compared to highly denatured chains and that a single-residue mutation can significantly alter unfolded-state dynamics and structure. This work suggests a view of the unfolded state in which surprisingly low diffusion rates could limit folding and opens the door for all-atom molecular simulation to be a useful predictive tool for characterizing protein unfolded states along with experiments that directly measure intramolecular diffusion.