J. Ponder, C. Wu, V. S. Pande, I. Haque, R. A. Distasio Jr., D. Lambrecht, M. Head-Gordon, G. Clark, M. Johnson, and T. Head-Gordon.
J. Phys. Chem. B., 114 2549-2564 (2010)
ABSTRACT.
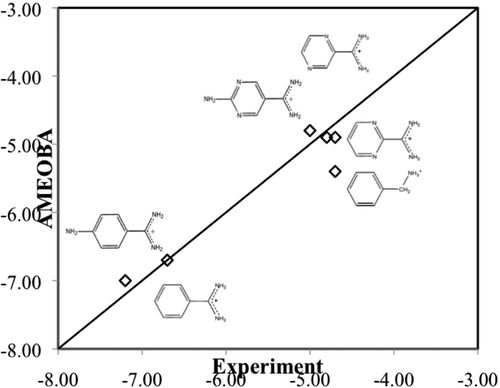
Molecular force fields have been approaching a generational transition over the past several years, moving away from well-established and well-tuned, but intrinsically limited, fixed point charge models toward more intricate and expensive polarizable models that should allow more accurate description of molecular properties. The recently introduced AMOEBA force field is a leading publicly available example of this next generation of theoretical model, but to date, it has only received relatively limited validation, which we address here. We show that the AMOEBA force field is in fact a significant improvement over fixed charge models for small molecule structural and thermodynamic observables in particular, although further fine-tuning is necessary to describe solvation free energies of drug-like small molecules, dynamical properties away from ambient conditions, and possible improvements in aromatic interactions. State of the art electronic structure calculations reveal generally very good agreement with AMOEBA for demanding problems such as relative conformational energies of the alanine tetrapeptide and isomers of water sulfate complexes. AMOEBA is shown to be especially successful on protein−ligand binding and computational X-ray crystallography where polarization and accurate electrostatics are critical.